Data Monitoring Committees in Practice
Subject safety in clinical trials has received much recent media attention, most publicly because of the death of a young man during a gene therapy study at the University of Pennsylvania.1 Some large and important clinical trials have been halted before their planned conclusions for safety and other concerns (for example, the Women's Health Initiative).2
Subject safety in clinical trials has received much recent media attention, most publicly because of the death of a young man during a gene therapy study at the University of Pennsylvania.1 Some large and important clinical trials have been halted before their planned conclusions for safety and other concerns (for example, the Women's Health Initiative).2



COLLAGE PAUL A. BELCI
Data monitoring committees (DMCs, sometimes called data and safety monitoring boards or DSMBs) have played an important role in providing oversight to critical clinical trials. A DMC is an independent group of experts who monitor unblinded safety and efficacy data while a trial is ongoing. They can recommend making changes to the conduct of the trial, including recommending to terminate the trial early for safety concerns, for overwhelming treatment efficacy, or for demonstrated futility in being able to show a benefit of treatment. In November 2001, the U.S. Food and Drug Administration (FDA) published draft guidance on DMCs3 explaining their need and how they should be integrated with clinical investigations. An extremely useful text by Ellenberg, Fleming, and DeMets covers many of the topics in this article.4
When is a DMC called for?
Not all clinical trials need a DMC. The draft guidance published by the FDA
3
provides some insight in deciding if a DMC is needed for a trial:
- DMCs have generally been established for large, randomized multisite studies that evaluate interventions intended to prolong life or reduce risk of a major adverse health outcome such as a cardiovascular event or recurrence of cancer. Because monitoring of accumulating results is almost always essential in such trials, DMCs should be established for controlled trials with mortality or major morbidity as a primary or secondary endpoint. They may also be helpful in settings where trial participants may be at elevated risk of such outcomes even if the study intervention addresses lesser outcomes such as relief of symptoms.
DMCs can also prove useful in clinical trials where mortality or major morbidity is not the primary endpoint. Figure 1 provides a diagram to help determine when a DMC is appropriate.4

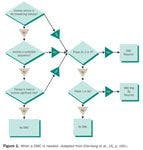
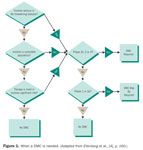
Figure 1. When a DMC is needed. (Adapted from Ellenberg et al., [4], p. 160.)
The DMC is in the unusual position of being able to review unblinded study data while the trial is ongoing. Using prespecified monitoring guidelines, the DMC might, based on the data it sees, recommend stopping a study early. In general, there are three scenarios under which a DMC might recommend stopping a trial:
- The data sufficiently demonstrates efficacy at such a high level of significance that it is very unlikely that if the trial were to continue the results would change
- There is a safety concern important enough to warrant changing or stopping the study
- There is sufficient data to indicate that even if the trial were to continue to its conclusion, it is extremely unlikely that the new treatment would show a statistically significant benefit (so-called futility curtailment or stopping for lack of efficacy).
Stopping a trial for futility can save subjects from unnecessary risk and can represent a substantial savings of the sponsor's money and time that would otherwise have been spent completing a trial highly unlikely to produce a positive result.
A DMC will always monitor for subject safety; however, the monitoring boundaries for overwhelming benefit or demonstrated futility must be specified to the DMC in advance, and are at the ultimate discretion of the sponsor with input from the DMC as well as the FDA or other regulatory agencies. Choosing appropriate monitoring boundaries is an important strategic decision for the sponsor. Furthermore, regulatory agencies should be informed about the use and responsibilities of the DMC and of the monitoring boundaries to be used.
As the DMC monitors for safety, it takes into account all available information. This might include, for example, the results from toxicology studies, or results from other clinical trials using the same class of drug. The DMC will need to carefully assess the risk-benefit balance. Because there are no set rules or guidelines for making decisions and recommendations regarding safety, this is an area where the expertise and judgment of the DMC members is critical.
The timing of DMC meetings is typically based on the number of patients with endpoint data; calendar time may also be a factor. DMC meetings might be convened based on some threshold, for example occurrence of more than one Serious Adverse Event, or more than two for a cancer trial since some level of side effects is tolerable. Again, the specifics of the disease and the treatment need to be weighed in setting up such guidelines.
The role of the DMC is advisory to the sponsor and study leadership. The sponsor (and possibly the study leadership) has ultimate responsibility for making decisions regarding the trial. As a trial is organized, it is important to consider to whom the DMC will report their recommendations. In many cases, a steering committee led by the sponsor is responsible for the conduct of the trial. Because the DMC may recommend major changes in the study, someone on the steering committee needs to have the authority to accept and implement the DMC recommendations.
Interim monitoring boundaries
Interim monitoring boundaries provide guidance to the DMC by defining the threshold beyond which the DMC might recommend stopping or altering a trial. They must be designed and agreed upon prior to the first "look" at the data. It is a nontrivial statistical task to design these boundaries, given that the DMC will review the data multiple times over the course of the study.
With repeated looks at the data, the observed p-values at each look cannot be compared using traditional measures of statistical significance (say, p < 0.05 or 0.01). The more often one looks at the data, the more likely one is to see a significant p-value totally by chance. Therefore, a higher level of statistical significance (smaller p-value) is needed earlier in the trial. An example of a possible boundary is shown in Figure 2. The "Normalized Z-Score" is a measure of the benefit or lack of benefit of the new treatment compared to the old (either placebo or standard) treatment. A larger Z-score indicates greater benefit, and a smaller (or negative) Z-score indicates lack of benefit or even harm. Thus, a Z-score of zero indicates neither benefit nor harm from the new therapy. If the observed Z-score crosses the top boundary of the "funnel" shown in Figure 2, then the DMC could recommend termination for overwhelming benefit. If the observed Z-score crosses the bottom boundary of the "funnel," then the DMC could recommend termination for demonstrated futility or lack of benefit.

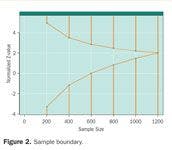
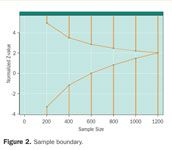
Figure 2. Sample boundary.
This example presumes that the maximum enrollment would be 1,200 participants and that the DMC will meet after every 200 participants to review data.
These boundaries can be modified to fit the desires of the sponsor, DMC, and regulatory agencies. The shape of the boundary determines how conservative the DMC will be in making its recommendations. For example, a sponsor may be pursuing multiple indications for a single agent. One indication will, potentially, have a larger market than another. The sponsor may design the boundaries for the less marketable indication so that it is more likely that the study would be stopped for lack of benefit (to cut losses sooner). Alternatively, the sponsor may want to collect as much data as possible. In this case, the sponsor would only want to stop the trial in the case of demonstrated harm to subjects, but would otherwise continue even in the presence of results that are highly positive or that have little chance of a final positive result.
The particular sample boundary shown in Figure 2 is an Emerson-Fleming symmetric boundary.5 An interesting feature of this particular boundary is that at the halfway point of 600 participants, a Z-score of zero or lower (no observed difference or a negative trend) would trigger a DMC recommendation to halt for demonstrated futility. If the trial went all the way to the end (n = 1200), the critical Z-score for statistical significance would be 2.02. This Z-score corresponds to a one-sided p-value of 0.022, very close to a traditional one-sided p-value of 0.025. This slightly higher hurdle at the end of the trial is indicative of the relatively small statistical price paid for the advantage of potentially stopping the trial with fewer participants.

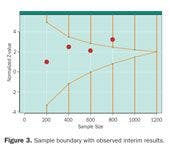
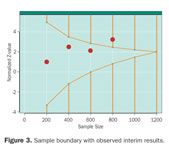
Figure 3. Sample boundary with observed interim results.
The statistics allow considerable flexibility in scheduling when the DMC will look at the results. For example, if the first scheduled analysis was after 200 subjects had been enrolled, but there was a safety concern that prompted the next analysis at 300 instead of 400 subjects, the boundaries could be adjusted to take this into account.
Figure 3 depicts a hypothetical trial with hypothetical results. At the first look (n = 200) there is basically no difference in the two treatments. Results look promising at the next two looks (n = 400 and 600), and at the fourth look (n = 800) the results have crossed the threshold for overwhelming benefit. Assuming there were no other issues that the DMC was concerned about (e.g., side effects that warranted additional monitoring), the DMC would issue a recommendation to terminate the trial early for benefit.
Setting up the DMC
A DMC is composed of independent experts who each bring separate knowledge to the group. The number of members can range from three to ten, with three to five being typical. A more complex study may require a greater number of DMC members. At least one DMC member will be a statistician and one will be the DMC chair. Clinicians knowledgeable about the disease indication should be represented, as well as clinicians knowledgeable in the fields of any major suspected safety effects (e.g., nephrology and cardiology). A few long, visible trials may include an ethicist or even a representative from a patient advocacy group.


Table 1. Sample DMC charter table of contents*
Locating potential members can be difficult. This is especially true in regards to identifying a DMC statistician. There are a relatively small number of statisticians experienced in looking at clinical trial data with the particular skill required to interpret early results of the trial.
DMC members are generally independent consultants to the sponsor. Their compensation should be fair, but not so excessive as to make an outside observer think that the DMC members may be unduly swayed to consider the sponsor interests over those of the subjects.
A DMC charter is a required3 document that defines the standard operating procedures of the DMC and the roles and responsibilities of all the involved parties. Some studies separate the DMC charter from the standard operating procedures for the DMC; others combine these two documents. A sample table of contents is given in Table 1.
Some publications have recommended using an "independent statistical center."6 This group receives data from the sponsor and the contract research organizations (CROs), as well as the randomization codes from the agency responsible for handling the central randomization or creating the master randomization list.
The independent statistical center is key to the DMC's functionality for a number of reasons. First, it ensures that the statistical report the DMC reviews can be created with the sponsor and the CROs remaining blinded to any treatment effect. By remaining blinded, the sponsor can alter the protocol or type of data being collected without suspicion that these changes were influenced by unblinded results. For example, if the sponsor were unblinded to the trial results, it would be unethical for them to make adjustments to the sample size if the expected difference between treatment arms was not as great as expected. Second, an independent statistical center can facilitate the logistics of the DMC that could unblind the sponsor, such as creating minutes of DMC deliberations or creating tables for the DMC.
The programming for the DMC report can be done by the sponsor using "dummy" randomization codes and then turned over to the independent statistical center to be merged with the actual codes. Alternatively, the programming can be contracted to the independent statistical center.
DMC meetings
The first meeting of the DMC will likely be organizational. While no data will be presented, the DMC will review and approve its charter. The sponsor can use this as an opportunity to explain the protocol to the DMC in some depth and also to give background on results from other related trials, for example detailing expected side effects of the treatment. The sponsor will also want to clearly communicate to the DMC its goals for the trial and any business objectives it wishes to be taken into consideration. This opportunity for a face-to-face meeting is especially valuable if some or all of the subsequent meetings are by teleconference.
Interim data and reports will be presented at subsequent meetings of the DMC. In preparation for these meetings, there are a number of steps and data sources that should be considered.
A timeline should be established well in advance of the DMC meeting. The timeline establishes the data cutoff for inclusion in the report, for sending data to the independent statistical center, and for sending DMC reports out for review.
The following are examples of the many sources of data that might be necessary for creating the DMC report:
- Traditional case report form data from the clinical database
- Serious Adverse Event (SAE) data from the safety database
- Randomization codes and up-to-date enrollment information
- Special assays or lab data that could unblind the sponsor
- Last-minute endpoint or mortality data prepared via an endpoint sweep.
Endpoint sweeps are a process for collecting up-to-the-minute primary endpoint and/or mortality information more current than CRF data. The DMC may want these data in order to make the best possible recommendation. For this reason, sponsors should plan for an endpoint sweep to be conducted within two weeks prior to the planned DMC meeting. Endpoint sweeps can be conducted using a log that the site completes either from information on hand or by telephoning the study participants.
In planning for "freezing" the data for a DMC, it is important to keep in mind that the DMC is generally concerned more about the completeness of primary and secondary endpoint and safety data than whether the data has outstanding queries. The DMC may be willing to review unmonitored data, unadjudicated endpoints, and other data from nontraditional sources in its quest for maximal, timely data. The sponsor or the sponsor's CROs should prepare for a special round of monitoring visits in preparation for a DMC meeting or should plan on the endpoint sweeps described above. Information on data completeness should be presented to the DMC.
Typically, two versions of the DMC report are produced: an open report that shows results aggregated across treatment arms (blinded data) and a closed report that shows results stratified by treatment arm (unblinded data). Only the open report goes to the sponsor, while both reports get sent to the DMC. These reports are distributed so that there is sufficient time, usually at least a week, for the DMC and sponsor to review their respective reports prior to the meeting.
A typical DMC meeting will begin with an open session where a sponsor representative will discuss the blinded data, trial management issues, or any other topics not involving unblinded data. The open session is an opportunity for the DMC to learn whether the sponsor's objectives for conducting the trial have changed. Has a competitor introduced a new drug? Have there been any interactions with the FDA? The open session is attended by the DMC, the sponsor, and possibly the sponsor's CROs.
The open session is followed by a closed session attended by the DMC and perhaps a representative of the independent statistical center. The DMC reviews the unblinded data, formulates a recommendation, and then the independent statistical center typically collects and destroys all copies of the closed report.
The DMC meeting finishes with another open session where the recommendations of the DMC are verbally given. DMC recommendations are communicated in writing a short time later. The audience of the DMC for this open session could be the whole steering committee, or a select few individuals from the steering committee. It should be stated in the DMC charter who can attend this session, so knowledge of the recommendation is initially limited to a small number of people, particularly if there will be a major change to the study. The sponsor can forward the DMC recommendations to institutional review boards (IRBs) upon request. An increasing number of IRBs are recognizing the value of independent DMC review of clinical trial data.
The DMC recommendations can be terse ("The DMC met on this date. The DMC recommended that the trial continue without change.") or they can be more involved. It is not uncommon for there to be minor recommendations about changing or adding statistical tables in the DMC report or encouraging the sponsor to reduce loss to follow-up, for example. Most of these recommendations can be easily handled. But the situation can get more complicated when the DMC comes out of its closed session recommending that the trial be terminated or undergo major changes.
Reacting to a major change recommendation
Unfortunately, it is rare that a DMC recommends a trial be stopped early for overwhelming efficacy. Most trials are halted due to lack of efficacy or due to a safety concern. In making the decision to stop or make a major change to a study, the DMC uses the statistical monitoring boundaries as specified in the charter, but must also use its expertise in evaluating a risk-benefit ratio using all available information. If the DMC decides that the risk-benefit ratio does not warrant continuing the trial, the sponsor should act promptly on the DMC recommendation.
In our experience, well over half of trials that use interim monitoring plans are stopped early. Therefore, study leadership should plan for the steps they will take should a DMC recommend a major change to the trial. Once the DMC has made a recommendation, the sponsor should be prepared to act upon that recommendation promptly and efficiently.
Some issues to consider when planning for such an eventuality are:
- Who should be notified if the DMC recommends a major change?
- Does that person or small group of people have the authority to accept or reject the recommendation and act on behalf of the sponsor?
- Prior to accepting the DMC recommendation, who if anyone from the sponsor or study leadership should review the unblinded results that led to the recommendation?
- How will the clinical investigators be notified?
- How will the sponsor be assured (for reasons of subject safety) that all subjects have been notified and that proper changes in therapy have been made?
- How will IRBs be notified?
- How will the FDA or other regulatory bodies be notified?
- If the sponsor is a publicly traded company, how will the financial market reaction be handled?
- Who at the sponsor should see unblinded study results prior to closing and locking the study database?
Who should be notified if the DMC recommends a major change? A small subcommittee of the study steering committee (sometimes called an executive committee) is usually charged with interacting with the DMC. This committee should include a representative of the sponsor with authority to act on behalf of the sponsor. In the event that the DMC makes a recommendation to stop or make a major alteration to the study, this group might need access to unblinded results in order to decide whether to accept the DMC recommendation. The executive committee will likely remain blinded to individual subject randomizations, but they may review the unblinded DMC reports and request additional analyses as necessary to clarify the issues that led to the DMC recommendation. The committee may also ask the DMC to review additional data as appropriate. One reason that this responsibility is limited to a small group is so that if the sponsor elects to continue the trial, as few people as possible have seen unblinded interim results. The sponsor may consult with regulatory agencies before deciding whether or not to accept the DMC recommendation.
How are clinical investigators and others notified of major changes? The urgency for notifying clinical investigators and study subjects depends on the issues that led to termination of the study. Certainly, if the DMC identified serious safety issues, the investigators and subjects need to be contacted as soon as possible. With global studies, disseminating DMC recommendations in a timely manner can present logistical challenges.
Again, depending on the reasons for termination and the nature of the treatment, subjects may be notified by telephone to discontinue their study medication. Some may need to see their clinical investigator or personal physician to make decisions about cessation of study treatment and subsequent therapy.
In the event of termination, IRBs and regulatory agencies need to be promptly notified. The sponsor has regulatory obligations to notify the FDA if it decides to terminate a trial. The safety of the subjects should be the primary concern in making decisions regarding study termination.
Conclusion
Employing a DMC to monitor a clinical trial is a powerful tool that enables the sponsor to remain blinded to the accumulating results while at the same time protecting subject safety and allowing for possible early termination of a clinical trial for benefit, futility, or safety concerns. The recommendations of the DMC are just that, recommendations. It remains the responsibility of the sponsor to make the ultimate decisions regarding the course of the trial.
References
1. L. Thompson, "Human Gene Therapy: Harsh Lessons, High Hopes," FDA Consumer, 34 (5) September-October, 2000; also at
http://www.fda.gov/fdac/features/2000/500_gene.html
.
2. J.E. Roussouw et al., writing group for the Women's Health Initiative Investigators, "Risks and Benefits of Estrogen Plus Progestin in Healthy Postmenopausal Women," JAMA, 288 (3), 321-333, 2002.
3. http://www.fda.gov/cber/gdlns/clindatmon.htm
4. S. Ellenberg, T. Fleming, D. DeMets, Data Monitoring Committees in Clinical Trials: A Practical Perspective (John Wiley & Sons Inc., New York, 2002).
5. S. Emerson and T. Fleming, "Symmetric Group Sequential Test Designs," Biometrics, 45, 905-923, 1989.
6. M. Fisher, E. Roecker, D. DeMets, "The Role of an Independent Statistical Analysis Center in the Industry-Modified National Institutes of Health Model," Drug Information Journal, 35 (1) 115-129, 2001.
David Kerr,* MS, is project director, Ruth McBride, BS, is chief technology officer, and Lynn Shemanski, PhD, is senior biostatistician with Axio Research Corporation, a division of Solutia Pharmaceutical Services Division, 2601 4th Avenue, Suite 200, Seattle, WA, 98121, (206) 577-0217, fax (206) 547-4671, email: davidk@axioresearch.com.
*To whom correspondence should be addressed.







What Can ClinOps Learn from Pre-Clinical?
August 10th 2021Dr. Hanne Bak, Senior Vice President of Preclinical Manufacturing and Process Development at Regeneron speaks about her role at the company as well as their work with monoclonal antibodies, the regulatory side of manufacturing, and more.





