A to Z Trial Integration
The last mile in clinical research is integrating the research site to create a complete information supply chain.
Related Reading
For more on managing clinical trial data in today’s high-tech environment, check out the following articles:
- Data Integration: Past and Future By Solomon Shacter
- Integrating EHR with EDC: When Two Worlds Collide By Paul Bleicher
- Multi-Trial Data Integration By Ralph Hughes
- Raising the Standards for Clinical Integration By Marie MacDonald
There are significant opportunities to improve current methods of electronic data capture (EDC) in clinical research for both government and industry sponsored clinical trials. Current methods generally require investigators to keypunch data into EDC systems individually supplied by sponsors for each trial. While this is an improvement over paper-based methods, the current EDC model falls well short of possible efficiency improvements and, in effect, reinforces work redundancy at investigator sites.
A far more effective approach would be for clinical investigators and sponsors to share a common system platform or, at least, for systems at investigator sites to be capable of directly supplying high-quality, auditable clinical trial data to sponsors and be in compliance with CFR Part 11.



(PHOTOGRAPHY: DIGITAL VISION, EYEWIRE, ILLUSTRATION: PAUL A. BELCI)
True integration
For some time now, forward-thinking investigators and sponsors alike have suggested that extracting clinical trial data directly from the electronic health record (EHR) would be far more efficient.
A thoughtful analysis of this opportunity, such as the March 2006 Applied Clinical Trials article "Integrating EHR with EDC: When Two Worlds Collide" by Paul Bleicher,1 rightly suggests skepticism about what's possible when EDC and EHR collide.
In short, integrating EHR and EDC is not viable today unless source EHR data is first fed into CFR Part 11 compliant systems for clinical trial fulfillment—which is exactly what some researchers have begun to do. The approach is conceptually straightforward: Use one system solution that at once addresses the needs of investigators and sponsors. Such systems and integrated supply chains are in production today, conducting trials electronically from source data to sponsor submission. The approach is very similar to supply chains commonly found in other industries such as manufacturing.
This article contains a description of how such fully integrated, clinical trial supply chains work from a systems perspective; the required systems technology; benefits for sponsors; supporting government and industry initiatives that are facilitating such integration; and some business model suggestions that will accelerate the evolution of integrated clinical research supply chains.
Last mile analogy
There is a need to solve "the last mile" problem in clinical research. The notion of the last mile is most often associated with the telecommunications industry, where wiring the last mile—the cable or phone lines that go from the street into the house—is among the most time-consuming and expensive system infrastructure challenges. Notably, in that industry such wiring is also necessary to deliver meaningful system value.
The analogy of the last mile also applies to clinical research, where the last mile problem is integrating research sites. Traditional EDC has gone part of the distance. To use the telecommunications analogy in the current model, investigators and their staffs using traditional EDC leave behind the systems and devices they use in their homes, walk to the bottom of their driveways or out to the sidewalk, log in to the system, enter trial data, and walk back into their homes and continue using whatever systems and devices they use on a day-to-day basis.
As such, clinical research fulfillment is a broken supply chain. Indeed, some would argue that the current state-of-the-art systems introduced increase workloads for many research sites, which in turn adversely impacts sponsor productivity and contributes to the extended time periods required to conduct clinical trials and obtain related drug/device approvals. Few would disagree that the current supply chain and EDC model likely perpetuates tens of millions of dollars annually in costs and delays in clinical research execution.
The only way to fix the supply chain is to solve the last mile problem. From an economic perspective, the constituency that stands to gain the most from a complete supply chain is research sponsors, particularly industry sponsors, simply because they generally have the most to gain economically from improved clinical trial execution. These gains in turn translate to better treatment options sooner for patients.
The system solution
The capabilities that need to be supported to enable an integrated supply chain include the following:
- The ability to support investigator and sponsor clinical trial information through one system platform.
- The ability to manage data across studies and patients without changing systems for each study.
- Integration capabilities so that individual consortium members can have source clinical data feeds, such as lab and demographics, from internal source systems to the EDC solution and on to the sponsor.
- The ability to broadcast clinical trials to investigators with otherwise uniquely configured research systems.
- A fully compliant FDA CFR Part 11 system validated by industry sponsors.
- A pure Internet platform solution that is flexible, highly configurable, and affordable.
A number of EDC solutions do a solid job with individual studies, but few vendors address all the above needs. An integrated system designed from the ground up to support all the above capabilities is even more compelling. Simply put, the most effective way to eliminate redundant data entry and other widely documented inefficiencies in investigator–sponsor interactions is for investigators to use a system on a daily basis that also serves the needs of sponsors (see Figure 1).

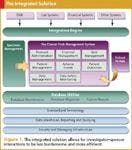
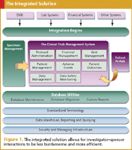
Figure 1. The integrated solution allows for investigatorâsponsor interactions to be less burdensome and more efficient.
The general system specifications required to address all of the above needs include:
- A system architecture that is both patient- and study-centric.
- A solution and system design that integrates study management via a clinical trials management system and EDC.
- Advanced technology to support federated databases and research grids—a must to enable investigator utility and address the clinical research last mile problem.
- A product that incorporates industry sponsor standards, such as Clinical Data Interchange Consortium (CDISC),2 as well as standards for government-sponsored trials, such as those of the National Cancer Institute's (NCI) Cancer Bioinformatics Grid (caBIG™) and hospital medical record system (HL7).3
- An integrated interface engine coupled with deep expertise and rich software libraries to support efficient, reliable EHR source data integration.
- Most or all the typical functions of a traditional EDC solution.
Steps to integration
For sponsors, the process of implementing studies in a fully integrated supply chain is similar to conducting a clinical trial using traditional EDC. While extensive pretrial operations are also supported, for purposes of this article, this discussion begins from the time the study protocol and case report forms are IRB approved and ready to be configured in the system.
Step 1: Configure. The first step is to configure the study in the system. The two general configuration activities are study set up and site participant set up. These activities are the same as those performed in a traditional EDC-based clinical trial.
As compared to some EDC solutions, one difference may be that the system needs to be easy enough so that investigator staff can set up trials without vendor or systems personnel. This is because that same system must be usable and affordable to support other clinical trials, such as investigator-initiated trials that have more limited budgets than most industry-sponsored trials. It follows that such tasks can also be easily conducted by sponsor or CRO personnel.
Step 2: Broadcast. In this step, called study broadcasting, the sponsor or its designate notifies site participants that the study is ready for patient accrual. Participating sites that already have the system receive the study in their local or vendor-hosted database. Authorized users at sites not already using the systems are given secure, on-line access to particular studies directly from the sponsor, CRO or lead investigator—very much like traditional EDC.
Step 3: Accrue and fulfill. Next, sites accrue patients and carry out the study. There are three kinds of sites from a systems perspective. Sites with source system interfaces fulfill corresponding portions of the study electronically (i.e., no manual data entry for electronically sourced data). Where source system interfaces are not implemented, trial-specific data for individual trials is posted from within the system environment in use at the site, leveraging data and workflow already built into the application. Patient registration and scheduling, for example, are fully integrated with EDC and trial fulfillment. New sites (sites not already using the system) post trial data in the same manner as traditional EDC.
Some investigators may adopt the system to manage clinical research across the department or enterprise and some may not. Either way, any sponsor-authorized investigator can participate in the clinical trial.
Communications and workflow management tools also are required. These capabilities enable sponsors and sites to interact and monitor trial activity throughout the trial life-cycle. With traditional EDC, the bulk of the value and time savings comes around the time of trial database lock. In an integrated clinical trial supply chain, process and workflow improvement and system-based investigator and sponsor communications can occur throughout the study life-cycle.
Step 4: Download. When study or case report form data is completed and authorized through e-signature, study data is downloaded to the individual study database, which may be hosted by the vendor. The sponsor, CRO, lead investigator site or coordinating investigator site could also host the database. As such, sponsors can have continuous, up-to-the-minute access to study status information throughout the trial life-cycle. Complete CFR Part 11 compliant audit trails and compliance controls must be in place throughout the study life-cycle.
Step 5: Resolve. A wide range of easy-to-use system and workflow tools should support the entire research fulfillment process, including alerts and reminders, query management tools, ad hoc reporting, audits trails, and other tools. These system features help sponsors promptly and efficiently review data, compare site performance, interact and communicate with sites, review results, resolve queries, and ensure complete, accurate data for subsequent trial database lock.
Step 6: Post and lock. The activities of posting trial data and supporting database lock are essentially the same as with traditional EDC. Data should be available in SAS, XML or any delimited sponsor specified format. In the case of trials sponsored by such large sponsors as the National Institutes of Health (NIH), study results can be posted directly to their trial databases.
Is it feasible?
Not only is it feasible, it's being done today on a production scale in a variety of research contexts by both investigators and sponsors. One group of 14 networked medical centers is electronically downloading complete trial data in a federated database environment (meaning some members have separate physical database instances) to a large sponsor on 46 trials.
Another group of large cancer centers in a similarly federated environment supports both industry- and government-sponsored clinical trials in prostate cancer. Among the centers in the group are Memorial Sloan-Kettering Cancer Center, UCSF Comprehensive Cancer Center, Dana-Farber Cancer Institute, Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, University of Michigan Comprehensive Cancer Center, and University of Wisconsin Comprehensive Cancer Center. Some members of this group source clinical trial data electronically; all submit electronically for both industry and government sponsors. The group and its individual members are running more than a dozen clinical trials (see Figure 2).

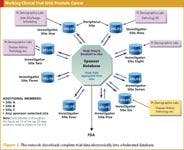
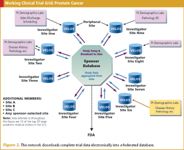
Figure 2. This network downloads complete trial data electronically into a federated database.
There are a number of other such networks where multiple sites and sponsors are collaborating in a federated, or secure, shared database environment, each with the fullest protection for their data because the data is aggregated only to that specific sponsor's database instance. More generally, my staff and I have regular exposure to organizations that receive about 30% of NIH extramural grant funding. Most of these customer organizations have multiple research organizations collaborating in clinical research using a common system platform; many that do not have plans to do so.
In terms of ease-of-use, some research networks, both investigator- and sponsor-initiated, have set up their own multisite networks and begun conducting research in as little as three weeks with only a few hours of Web-based training. This is not an average. The process generally takes longer and additional training should be encouraged, but it does demonstrate ease-of-use and the opportunity for investigators and sponsors to expediently assemble and disband for research projects.
Advantages for sponsors
Many research sites immediately recognize that this integrated, less-redundant approach saves time and money. The same is true for sponsors. As compared to traditional EDC, clinical trial sponsors can achieve cost savings in the form of reduced monitoring costs, fewer queries, fewer data entry mistakes, reduced site training requirements, less paper-source documentation verification, lower costs to implement trial protocol changes, and lower unit costs to collect given quantities of data from each site (see Figure 3).

Figure 3. EDC capabilities go far beyond data capture, and should be used throughout the trial to maximize productivity.
More importantly, clinical trial sponsors benefit from all the above in the form of reduced elapsed time, and such benefits accrue throughout the clinical trial fulfillment life-cycle—not just at database lock.
- Less monitoring takes less time.
- Fewer queries take less time.
- Fewer data entry mistakes require less time.
- Fewer system training needs accelerates data turnaround.
- Protocol changes, if needed, require less time.
- Manually transferring data from source documents to case report forms takes less time.
Additionally, the advantage of the research supply chain increases with study volume. In the traditional EDC approach, each new study is started afresh. In the integrated supply chain, sponsors simply broadcast new studies directly into the investigator "homes."
Numerous other benefits result from additional and timely exposure to what's happening at the sites (such as patient accruals and study status); from opportunities to develop closer working relationships and multistaged research programs with leading investigators; and from opportunities for positive change and continuous quality improvement that naturally emerge from an integrated supply chain.
Supporting initiatives
Software vendors, investigators, and sponsors have been successful implementing complete clinical research supply chains in part as a result of certain government initiatives, most notably caBIG™ and the Clinical and Translational Science Awards (CTSA). Continued industry-sponsored improvements in data exchange standards like CDISC and efforts to marry standards, particularly CDISC and HL7, will also create opportunities over time for far more substantial sponsor–investigator integration.
caBIG™ is the NCI's initiative to improve data exchange standards; drive research collaboration and information sharing among investigators; and improve system infrastructure for cancer research at investigator sites. A $25 million, three-year pilot project, which concludes this year, caBIG™ has succeeded in aligning research centers, technology vendors, and government to address some deeply rooted, systemic inefficiencies in the conduct of cancer research in the United States.
As with all pilots, some aspects of the program have been more successful than others. One of the successes has been the emergence of vendors supporting adoption of state-of-the-art research systems at many U.S. medical centers. caBIG™ has been closely watched by other NIH and similar organizations internationally. Accomplishments and lessons learned in caBIG™ will likely be applied beyond cancer research. This improved clinical research infrastructure is already benefiting sponsors today and will ultimately benefit patients.
The CTSA is a new government program intended to foster sweeping changes in our national research system infrastructure, most notably by significantly increasing focus on translational science (i.e., the activity of converting discoveries in basic science into viable therapies or, in industry parlance, from bench-to-bedside). The CTSA program is galvanizing innovation among medical centers, particularly academic medical centers, in developing improved infrastructure to support translational science, which largely means clinical research infrastructure. As such, a direct result of the CTSA program is likely to be the adoption of far more advanced systems to support clinical research, which in turn benefits sponsors.
System solutions such as the one described in this article are particularly well suited to support the needs of CTSA recipients, their sponsor beneficiaries, and similar initiatives under way internationally.
When it comes to standards, the comparison of clinical research to telecommunications and manufacturing supply chains is actually unfair. Medicine in general, and clinical research specifically, have far more complex data interoperability needs. The continued progress made by CDISC in defining information standards to enable systems-level communication for clinical research will accelerate evolution of integrated clinical research supply chains.
For purposes of automating clinical source data feeds, a priority for CDISC is the marriage of CDISC and HL7. Anyone closely familiar with the issues here understands that this is no simple marriage. The reasons for this are many, and include the fact that HL7 has many flavors, some of the standards are still overly broad for robust use, and adoption at investigator sites for purposes of electronic source documentation will take time (hint: implementing EHR is the horse, research data exchange standards are the cart).
Finally, as clinical research systems professionals, we have to remember that our thought leaders are clinicians with therapies in continuous change. They're also researchers who are going to identify new ways of classifying data. Some experts would argue that there will never be universal standards.
As relates to data exchange standards, we're going to have to settle for partial wins. At least we can automate lab data feeds, for example, for studies that are lab intensive, and for any particular clinical trial, we can agree on one or another set of standards and combinations therein. What's important from a systems perspective is that the technology can easily adapt and support common, individual data standards for given trials. To solve the last mile problem, this has to be done without requiring investigators and sponsors alike to start afresh on new system platforms each time. This in turn necessitates that federation occurs across separate physical databases. Such technology is available and in production now.
The connected community
Today, research sponsors and investigators alike are forming, disbanding, and reforming clinical trial networks on a common system platform. The key difference between such systems-based communities and traditional EDC networks is the high level of system and process re-use that becomes feasible.
If addressing the functional needs of both investigators and sponsors is critical, addressing the security and database requirements of each institution is even more so. Investigator A needs access to data that Investigator B cannot access. Sponsors need to be sure they cannot under any circumstances access patient identifiable data. And it goes without saying that Sponsor A's patient-identifiable data cannot reside in the same physical environment as Sponsor B's data. The only way to address these security needs while also solving the last mile problem is through federated databases.
A federated database is one where individual "members," each with a discrete database, agree to abide by common (federal) rules under certain circumstances, most notably for purposes of executing any particular clinical trial, while continuing to operate under separate rules covering their local implementations (or nation/states).
In addition to technology, some business model choices can be made that will foster greater collaboration, supply chain integration, and investigator–investigator and sponsor–investigator collaboration. These include:
- Licenses without organizational boundaries. If system licenses are not restricted by organization boundaries, users can create their own research networks, extend research into their local, state, national or international communities, and interact with other networks, enabling a network of research networks.
- Content sharing. When member organizations voluntarily share system content, such as workflow, case report forms, publicly sponsored clinical trial protocols, etc., and vendors provide customers the technology to enable such sharing, individual member workloads can be significantly reduced.
- Free or low-cost study broadcast licensing. A business model (particularly for nonprofit, investigator-initiated trials) that allows members to broadcast trials to members and nonmembers at a low cost or for free will facilitate natural supply chain evolution.
- Public service. Continued support from many vendors for government and industry initiatives such as the NCI's caBIG™ and CDISC, with an emphasis on practical solutions, will serve the clinical research community well.
Summary
As an industry, we have a good distance to go before fully integrated supply chains become the norm. Together we're operating under various constraints, some of which can be removed, as this article demonstrates, and some of which are hard to control, most notably the pace of EHR adoption.
Researchers have demonstrated the potential to transform otherwise discontinuous clinical research information supply chains to ones that are more integrated and effective for investigators and sponsors. These achievements create an opportunity not just for incremental improvements, but for fundamental and pervasive improvements in clinical research productivity and efficiency. Automated source data is one of numerous opportunities for improvement. Those forward-thinking clinicians and researchers may not be so far off after all.
John McIlwain is president and chief executive officer of Velos, Inc., 2201 Walnut Avenue, Fremont, CA 94538, 510-580-2660, email: jmcilwain@velos.com
References
1. P. Bleicher, "Integrating EHR with EDC: When Two Worlds Collide," Applied Clinical Trials, March 2006 Supplement, http://www.actmagazine.com/appliedclinicaltrials/article/articleDetail.jsp?id=310798.
2. http://www.cdisc.org/about/index.html.
3. https://cabig.nci.nih.gov/.









FDA Fast Tracks Johnson & Johnson’s Nipocalimab for Fetal Neonatal Alloimmune Thrombocytopenia
March 27th 2024Johnson & Johnson is moving forward with a pair of Phase III trials of nipocalimab to reduce the risk of fetal neonatal alloimmune thrombocytopenia in alloimmunized pregnant patients.


Citius Pharmaceuticals Resubmits BLA to FDA for Lymphir to Treat Cutaneous T-Cell Lymphoma
March 19th 2024Pivotal Phase III Study 302 trial data show an objective response rate of 36.2% based on an independent review committee assessment in the treatment of relapsed/refractory cutaneous T-cell lymphoma.




