Initial Design Considerations for Immuno-Oncology Trials
Perspectives and insights regarding regulatory considerations when planning and conducting immunotherapy studies.



Over the years, KCR Trial Execution and Consulting has assisted in conducting and executing clinical trials in immuno-oncology around the globe for biopharmaceutical and biotechnology clients. Valuable, useful perspective and insights have been gained through these interactions with regulatory authorities in the European and U.S. market, which are corroborated by reviewing current and relevant guidance and literature. We would like to share these experiences with our drug development and clinical researchers, as well as industry peers.
Comparisons between traditional drug trials and immuno-oncology studies
1. Traditional early Phase I/II drug trials. Traditional clinical trials on cytotoxic drugs have followed a Phase I/II/III transition model, with a typical 3+3 designs in dose escalation and expansion in early clinical trial stages.1 Within this drug development framework, in the early phase of the clinical studies, i.e., the Phase I or “first in human” study, the focus is to determine an appropriate drug dose based on toxicity profile of the investigational agent. The dose finding experiments are usually designed with some preclinical and/or animal data to determine the dose levels of the drug. During this time, the so called investigational medicinal product (IMP) is first administered in a cohort of three unselected subjects; each drug level is used in no more than six patients of two cohorts. The rule of thumb is if the first three subjects have not encountered any drug-related toxicities, the drug concentration for the next cohort of three subjects would be escalated to a higher level. If toxicities are observed in only 1/3 of cases, the same drug level could still be applied in an additional three subjects. However, if the toxicities are observed in 2/3 or more of the subjects, which is identified as dose-limiting toxicity (DLT) and indicates the toxicity maximum tolerated dose (MTD) may have been exceeded.2
In general, in a Phase I study, no more than 30-50 patients with a prespecified stepwise dose escalation scheme are enrolled. The working definition of MTD is based on G3/4 drug-related toxicity observed in excess of 25%-33% of subjects. The RP2D recommendation is identified and derived from MTD.3
During the Phase II study, the emphasis remains on drug-related toxicities, however, efficacy is a primary consideration. Study endpoints, such as overall response rate (ORR) and duration of response (DOR) are introduced. The subjects to enroll for the Phase II trial may range from several hundred in dose-expanding cohorts of multiple arms, with or without active controls and randomization. The Phase II trials sometimes are divided as pilot (Phase IIa) and pivotal (Phase IIb) trials. The former trial is usually deployed to investigate efficacy and safety in a selected population, and the latter is a well-controlled trial to collect preliminary efficacy and safety data for planning a large scale Phase III clinical study forgo and no-go decision-making.3,4
Therefore, a working definition of MTD is critical for designing a early phase of clinical studies of cytotoxic agents, especially for dose-escalation and expansion cohorts. We also need to work out a case definition for inclusion and exclusion criteria for timely and efficient patient enrollment. Some surrogate indications of efficacy, such as ORR and DOR, are employed in Phase II trials for decision-making toward later phases of the clinical trials with respect to carrying out a registrational or Phase III trial.5
2. Characteristics of immunotherapy. In recent years, we have witnessed tremendous success in immunotherapy in cancer treatment where innovative therapeutic biologicals have been introduced into clinical practices for patient management of malignant neoplasms, including co-stimulatory biologicals, which include adoptive T-cell therapy such as tumor-infiltrating lymphocytes (TIL) and chimeric antigen receptors (CAR) T cells; as well as co-inhibitory treatment modalities such as the immune-checkpoint inhibitors. In contrast to cytotoxic drugs, there are several, notable characteristics in pharmacokinetics/pharmacodynamics(PK/PD) of the molecular entities in immuno-oncology which should be kept in mind when designing clinical trials.
Unlike the cytotoxic agents for cancer chemotherapy, new generation of the biologicals in immunotherapy are not the same in terms of their respective mechanisms and pathways as traditional chemotherapeutic agents, which are, first, not acting directly on malignant cells. The biologicals in immunotherapy are either affecting on innate and adaptive immune regulatory pathways or stimulating and strengthening immune functions or preventing tumor cells’ escape of immune surveillance in the human body.5
Furthermore, the therapeutic agents in immunotherapy are biologicals in nature; it has been observed that the dose-response of the immunotherapeutic agents do not follow the same PK/PD patterns of that of the chemotherapy, although the Cmax and AUC are dose-dependent. Most notably, the dose-response curve is largely non-linear. For example, it has been reported that median target occupancy of PD-1 and PD-L1 receptors of nivolumab and an anti-PD-L1 antibody is 64%–70%, which was the same for both the anti-PD1/-PD-L1 molecules and from doses ranging from 0.1 up to 10 mg/kg. However, the duration to achieve the minimal target modulation with such a wide range of the target occupancy remain elusive. Thereby, the dosing requirements and schedules to establish and maintain a sustainable target immunomodulation for effectiveness of the lasting therapeutic effect are often unclear.
The dosing scheme and regimen in combinations need to be ascertained and carefully justified based on limited chemistry, manufacturing and controls (CMC), or other preclinical data, and sometimes someone must simply rely on rationales of mechanistic perspective of target engagements.5
Lastly, the immune-related adverse events (irAEs) are of early and late onset.6 The early onset irAEs could be inflammatory reactions to the immunotherapeutic treatments such as hepatitis, enterocolitis, hypophysitis, and nephritis, as well as more seriously tumor lysis syndrome, neurologic toxicity, and cytokine release syndrome, etc. Among the late-onset ones are most commonly fatigue, extensive rash or itching, diarrhea with blood or mucus, or severe abdominal pain, weight loss, nausea/vomiting, excessive thirst or appetite, excessive and/or frequent urination, shortness of breath, cough, etc. The late onset irAEs could occur as late as 10 to 16 weeks after the treatment initiation. In the context of observation of DLT, the late onset of the irAEs is what we need to consider.6
Initial design considerations for immuno-oncology
1. Study formulation and concept
Hypothesis and case definition. A clearly stated null hypothesis and case definition are the important first step in conceptualizing and formulating a clinical trial. For any oncologic clinical trial, case definition is an even more important element in a sense that cancer is not a single disease entity with a diversified molecular subtypes (heterogeneity) and would likely respond differently to treatments. An unequivocal case definition will facilitate clinical investigators to enroll target patients into a study through inclusion and exclusion criteria. Also, patients who were previously treated unsuccessfully are frequently encountered in a variety of different clinical environments. These patients treated with biologicals targeting alternative pathways who failed could still be eligible or manageable by immunotherapies in a second or third line of treatments. Under such circumstances, a procedural case definition will be helpful. A clearly stated case definition if being standardized will be useful not only to understand nature of irAEs and to improve the recognition and management of common or serious irAE associated with I/O studies, but also facilitate study execution, data analysis, and interpretation.6-8



DLT for MTD determination. The primary goal of the early phase to identify MTD through DLT, which is usually based on the percentage of occurrences of NCI Common Terminology Criteria (NCI CTCAE) G3/4 events. In traditional dose-escalation scheme for cytotoxic agents, the MTD is defined as that DLTs appear in one-third of the cases, which may not be applicable in IMP for immunotherapy with the 3+3 design, since the dose-response relationship is not straightforward or may not be found. Under such circumstances, DLT definition is critical for MTD identification. It is not uncommon that DLT for biologicals in immunotherapy may never be reached in any given trial in immuno-oncology. In recent years, maximum administered dose (MAD) is adopted instead of further investigations. In other trials, clinical researchers advocate for the use of biological optimal dose (BOD) in immunotherapy clinical development, which requires mathematical modeling skills.5,9-10
In addition, it pays to be mindful of the half-life of the biologicals (usually three to four weeks) when designing a study protocol with I/O therapeutic agents. Clinical researchers need to consider the very nature of I/O agents with delayed toxicity, in addition to estimating the assessment period of the DLT for MTD of an IMP in immunotherapy trials. It has been proposed that it should last five times of the half-life of the biologicals or longer, which is derived from preclinical studies and may extend beyond the first cycle of the dose escalation.10-13
Lastly, for combination regimens, the toxicity profile should be determined individually through a dose-range study, and for DLT, it could employ an accelerating titration design with the agent of a well-known toxicity profile and order if overlapping toxicities are expected. Since the current guidelines remains primarily for cytotoxic and targeted therapeutic drugs, the MTD determination is likely adjusted by integrating all available data, including PK/PD and toxicity profiles.5,10, 14-17
Biomarker enrichment, stratifications, and randomization scheme. IMPs in I/O are usually targeting immunological pathways for functional modulations to deliver therapeutic effects. Biomarker-driven designs for patient enrollment are desirable if a predictive biomarker is available for triaging potential candidates. Randomization schemes follow the positive and negative results of the biomarker testing for all comers, either in interventional or control arm, thereby the trial results can be interpreted later with confidence.17 There are many sophisticated molecular assays available nowadays on multiple platforms. However, the assays used for patient selection should be prospectively and independently validated.18
Combinations in regiment. Combination regimens for biologicals in immunotherapy have become routine practice. They have been adopted as a first-line or subsequent treatment strategy to improve efficacy of anti-cancer therapeutics. Various combinations of different therapeutic modalities are increasingly encountered in clinical trial settings. There is not a single strategy for trial design in drug combinations that can offer superior performance compared to the others in trial outcomes. The rationales for regimen combinations should be justified based on biological mechanisms of molecular mechanisms of immune modulations and targeted pathways, as well as other animal data from the preclinical studies. The relevance of animal data is also important since the activation of immunological reactions between different species could be of different thresholds in activation.19-20
Endpoint for outcome assessment. With the complex innovative designs (CID) for immunotherapies, some early phases ofclinical trials for biologicals in I/O have adopted, besides MTD, some intermediate efficacy endpoints such as minimum effective dose and objective response rate (ORR). However, ORR is considered a surrogate endpoint for overall survival (OS). Others include progression-free survival (PFS) and duration of response (DoR). For early-phase trials of immunotherapies with adaptive designs following master protocols, the efficacy endpoints are also deployed in dose escalation and expanding cohorts for early decision-making for phase transition or study design modifications, if necessary. The endpoints may be specific to therapeutic agents and their clinical benefits with respect to the effect size, which should be clinically meaningful and justifiable accordingly.21-24
2. Modeling for simulation
Design features and associated parameters can be simulated to optimize the study design configurations by using a variety of parameters and combinations to estimate a design for its operation characteristics. The input parameters could include the accrual rate, event rate, time to event, and effect size—thereby to provide the clinical investigators with some fundamental understanding with respect to the potential trial results. The computational approach is usually carried out with Monte Carlo simulations, leading to possible optimization of the study design in dose finding for early stages of clinical studies. Furthermore, modeling and simulations could also facilitate enrollment planning in feasibility study.25
3. Master protocol for adaptive trial design
Current business models of cytotoxic drug development have been outdated for IMP development in immunotherapy. Traditionally, the drug is following a trajectory path of sequential testing scheme, from Phase I to II, and efficacy and toxicity, later if data are supportive to Phase III trials. However, the biologicals in immunotherapy are highly complex and less predictable in PK/PD as the cytotoxic agents are. This requires trial planning and protocol designs being more flexible and agitative, which could respond to and integrate new data and new information from interim analysis, and, therefore, help to meet the challenges in biological complexity, restraints in study time, and requirements in cost containments.5,10
I/O trials are an appropriate setting to adopt an adaptive design with a master protocol for multiple arms because of the complexities and uncertainties associated with I/O therapeutic agents. The master protocols are of three types depending on the clinical trials, i.e., platform, basket, and umbrella. The platform master protocol is for trials of multiple immunotherapeutic biologicals of a single disease entity. The basket master protocol is employed in a trial for multiple diseases. The umbrella trial, while on the other hand is also a protocol for multiple therapies in a single disease, it is used usually for biomarker-driven trials and enrolls patients of certain biological characteristics in a specific trial with further randomization.26-28
Of note, master protocol is appropriate for clinical studies with adaptive designs and biomarker-defined subgroups. Thereby it should be applicable in I/O agent development.Other potential and major advantages of the adaptive study designs are the possibility of integrating new information continuously, such as that derived from real-world data, for dose selection and sample size reestimates, and thereby would be more appropriate in IMP development with an iterative development process, particularly in immunotherapy. Master protocols also support trial designs combining early phase trials such as Phase I/II trials for early efficacy determination through controlling for type 1 error. Both FDA and the European Medicines Agency (EMA) recognize that adaptive design has the potential to shorten development timelines and allocate limited resources more efficiently in meeting the requirements of scientific and regulatory guidelines.25, 29-31

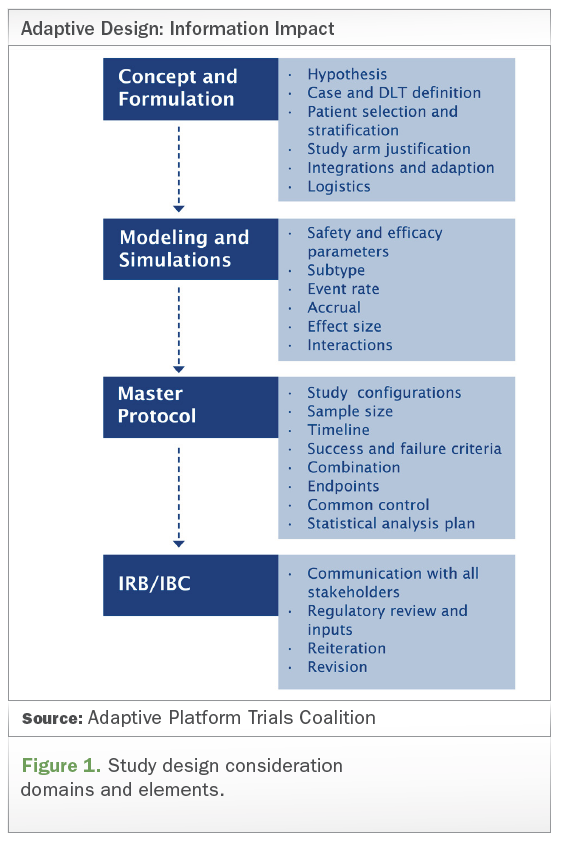
Click image to enlarge
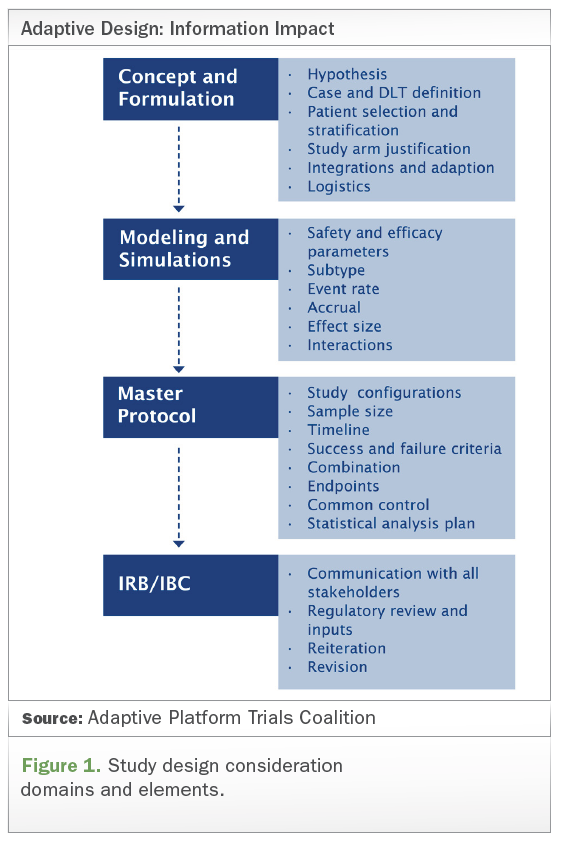
Other elements of the master protocol that should be taken into consideration include common control, success and failure criteria, study configurations, sample size, timeline, combination, endpoints, and statistical analysis plan, etc. (see Figure 1 above).
4. Planning, qualification process, and regulatory interactions
Medicine is a regulated business. To increase the odds of successful clinical development, it is imperative to keep the end in mind. That requires a well-planned process development and transparent communications with regulatory agencies. Both FDA and the EMA have established early interaction programs for the biopharmaceutical industry. The EMA qualification process addresses innovative drug development methods and tools, including presubmission consultation and advice; review by the scientific advice working party on novel methodologies of biomarkers, preclinical models, clinical outcome, modeling and statistics, etc. Similarly, FDA has also instituted the same qualification process tools to facilitate clinical development. It consists of three program—animal model, biomarker, and clinical outcome assessment.32-34
Although qualifying is not mandatory, it is advisable that sponsors interact with regulatory agencies early if there are issues in study designs that require regulatory opinions and feedback and/or in cases of further clarifications on agency guidance where documents are warranted.
Institutional review board (IRB) and institutional biological safety committee (IBC). Biosafety is a major concern in planning and conducting gene therapy and vaccine clinical studies. These studies may involve viral vectors, especially if they have a replication competent. The environmental and health impact should be carefully evaluated and follow biosafety guidelines in handling the sampling, storage, disposal, and administration of the recombinant nuclear acid materials, and developing appropriate protocols in management of patients in such a clinical trial.
IBC review and approval are required, as per a recently released guideline by the U.S. National Institutions of Health (NIH). The institutions and respective IBCs subject to the NIH guidelines are expected to establish policies and procedures to ensure that the research is conducted in full conformity with the provisions of the guidance. But any required documentation for approval is at the discretion of the institution and IBC. Sponsors should request IBC review and approval, or a central IBC, before or at the same time the clinical trial protocol is submitted to the IRB.35-36
In the EU, advanced technology medicinal products (ATMPs) are under the purview of the EMA and follow ATMP legislations and relevant regulations. Under the current GMO regulations, ATMPs are required to be evaluated for environmental and health risk either in the framework of deliberate release (Directive 2001/18/EC) or contained use (Directive 2009/41/EC) before or in parallel with the clinical trial application. The contained use is defined by the risk class of the GMOs and protective barriers by design to limit the unexpected exposure of potentially infective agents and harmful effects. In contrast, the deliberate release is in consideration of less restrictive measures to protect or limit the public from exposure in the general environment.37-40



Summary
IMPs in immunotherapy are different biochemical entities and highly complex in mechanistic dynamics, leading to less predictable behaviors in PK/PD and affecting their efficacy and toxicity profiles in vivo. Accordingly, the clinical development of IMPs for immunotherapeutics require a different mindset, fresh perspective, effective strategies, meticulous planning, innovative designs, and precise execution in order to accelerate the process of biological drug development in a timely and manageable manner.
References
- Cook N, Hansen AR, Siu LL, Abdul Razak AR. Early phase clinical trials to identify optimal dosing and safety. Mol Oncol. 2015 May;9(5):997-1007.
- Paoletti X, Ezzalfani M, Le Tourneau C. Statistical controversies in clinical research: requiem for the 3 + 3 design for phase I trials. Ann Oncol. 2015 Sep;26(9):1808-1812. doi: 10.1093/annonc/mdv266. Epub 2015 Jun 18. Review.
- Friedman LM Furberg CD, DeMets DL. Fundamentals of Clinical Trials 4thEds. Springer 2010.
- Liu MB and Davis K (eds.) A Clinical Trial Manual from the Duke Clinical Research Institute Lessons from a Horse Named Jim 2ndEdition. Wiley-Blackwell 2010
- Postel-Vinay S, Aspeslagh S, Lanoy E, Robert C, Soria JC, Marabelle A. Challenges of phase 1 clinical trials evaluating immune checkpoint-targeted antibodies. Ann Oncol. 2016 Feb;27(2):214-24.
- Curran C, Sharon E. Report on the FDA-AACR Immuno-oncology Drug Development Workshop. Cancer Immunol Res. 2017 Apr;5(4):282-285.
- EMA/CHMP/44762/2017 Committee for Human Medicinal Products (CHMP) Guideline on multiplicity issues in clinical trials https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-multiplicity-issues-clinical-trials_en.pdf
- FDA Guidance for Industry E9 Statistical Principles for Clinical Trials https://www.fda.gov/media/71336/download
- FDA/AACR Immuno-Oncology Drug Development Workshop, October 13 & 14, 2016 Washington DC https://www.aacr.org/wp-content/uploads/2019/11/I-O-Day-1-SlideDeck-safe.pdf
- Wages NA and Chiuzan C et al. Design considerations for early-phase clinical trials of immune-oncology agents. J Immunother Cancer. 2018 Aug 22;6(1):81.
- Coleman RL. Innovations in Immune Oncology Combination Clinical Trial Designs. https://www.fda.gov/media/114627/download
- Sheng J and Srivastava S, et al. Clinical Pharmacology Considerations for the Development of Immune Checkpoint Inhibitors. J Clin Pharmacol. 2017 Oct;57 Suppl 10:S26-S42.
- Eudocia Q Lee and Michael Weller et al. Optimizing Eligibility Criteria and Clinical Trial Conduct to Enhance Clinical Trial Participation for Primary Brain Tumor Patients. Neuro Oncol 2020 May 15;22(5):601-612.
- Cadth Technology Review:Optimal Use of 360 Report-Dosing and Timing of Immuno-Oncology Drugs https://www.cadth.ca/sites/default/files/ou-tr/ho0008-dosing-timing-immuno-oncology-drugs.pdf
- EMA Strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products https://www.ema.europa.eu/en/strategies-identify-mitigate-risks-first-human-early-clinical-trials-investigational-medicinal
- Shen J and Swift B et al. Design and Conduct Considerations for First-in-Human Trials. Clin Transl Sci. 2019 Jan;12(1):6-19. doi: 10.1111/cts.12582.
- Korn EL and Freidlin B. et al Adaptive Clinical Trials: Advantages and Disadvantages of Various Adaptive Design Elements. J Natl Cancer Inst. 2017 Jun 1;109(6).
- Mehnert JM and Monjazeb AM et al. The Challenge for Development of Valuable Immuno-oncology Biomarkers. Clin Cancer Res. 2017 Sep 1;23(17):4970-4979.
- Morrissey KM and Yuraszeck TM, et al. Immunotherapy and Novel Combinations in Oncology: Current Landscape, Challenges, and Opportunities. Clin Transl Sci. 2016 Apr;9(2):89-104.
- A K S Salama AKS and Moschos SJ, Next Steps in Immuno-Oncology: Enhancing Antitumor Effects Through Appropriate Patient Selection and Rationally Designed Combination Strategies. Ann Oncol 2017 Jan 1;28(1):57-74.
- Anagnostou V and Yarchoan M et al. Immuno-oncology Trial Endpoints: Capturing Clinically Meaningful Activity. Clin Cancer Res. 2017 Sep 1;23(17):4959-4969.
- FDA Multiple Endpoints in Clinical Trials Guidance for Industry DRAFT GUIDANCE https://www.fda.gov/media/102657/download
- FDA Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics Guidance for Industry DECEMBER 2018 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-trial-endpoints-approval-cancer-drugs-and-biologics
- EMA Clinical efficacy and safety: antineoplastic and immunomodulating agents. https://www.fda.gov/media/71195/download
- Adaptive Platform Trials Coalition. Adaptive platform trials: definition, design, conduct and reporting considerations. Nat Rev Drug Discov. 2019 Oct;18(10):797-807.
- FDA GUIDANCE DOCUMENT Master Protocols: Efficient Clinical Trial Design Strategies To Expedite Development of Oncology Drugs and Biologics Draft Guidance for Industry OCTOBER 2018 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/master-protocols-efficient-clinical-trial-design-strategies-expedite-development-oncology-drugs-and
- EMA European regulators’ view on platform trials Lessons recently learned https://www.efspi.org/Documents/Events/Events%202018/3rd_RegStat/32%20Hofner.pdf
- FDA Adaptive Designs for Clinical Trials of Drugs and Biologics Guidance for Industry https://www.fda.gov/media/78495/download
- European Medicinal Agency (EMA): Point to Consider on Methodological Issues in Confirmatory Clinical Trials with Flexible Design and Analysis Plan. The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Human Use. CPMP/EWP/2459/02, London, UK 2002.
- European Medicinal Agency (EMA): Reflection paper on Methodological Issues in Confirmatory Clinical Trials with Flexible Design and Analysis Plan. The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Human Use. CPMP/EWP/2459/02, London, UK 2006.
- EMA Guidance for companies considering the adaptive pathways approach https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/guidance-companies-considering-adaptive-pathways-approach_en.pdf
- FDA GUIDANCE DOCUMEN Qualification Process for Drug Development Tools Guidance for Industry and FDA Staff DECEMBER 2019 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/qualification-process-drug-development-tools-guidance-industry-and-fda-staff;https://www.fda.gov/drugs/drug-development-tool-ddt-qualification-programs/drug-development-tool-qualification-process-transparency-provisions
- FDA Guidance for Industry and Review Staff Target Product Profile - A Strategic Development Process Tool DRAFT GUIDANCE https://www.fda.gov/media/72566/download
- EMA Qualification of novel methodologies for medicine development https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-advice-protocol-assistance/qualification-novel-methodologies-medicine-development
- FDA Determining the Need for and Content of Environmental Assessments for Gene Therapies, Vectored Vaccines, and Related Recombinant Viral or Microbial Products Guidance for Industry https://www.fda.gov/regulatory-information/search-fda-guidance-documents/determining-need-and-content-environmental-assessments-gene-therapies-vectored-vaccines-and-related
- NIH GUIDELINES FOR RESEARCH INVOLVING RECOMBINANT OR SYNTHETIC NUCLEIC ACID MOLECULES (NIH GUIDELINES) APRIL 2019DEPARTMENT https://osp.od.nih.gov/wp-content/uploads/NIH_Guidelines.pdf
- Salmikangas P1,2, Schuessler-Lenz M et al. Adv Exp Med Biol. 2015;871:103-30. doi: 10.1007/978-3-319-18618-4_6. Marketing Regulatory Oversight of Advanced Therapy Medicinal Products (ATMPs) in Europe: The EMA/CAT Perspective.
- EMA/CAT/852602/2018 Committee for Advanced Therapies (CAT) Guideline on quality, non-clinical and clinical requirements for investigational advanced therapy medicinal products in clinical trials Draft (EMA 1 31 January 2019) https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-quality-non-clinical-clinical-requirements-investigational-advanced-therapy_en.pdf
- Belgian Biosafety Server:EU regulatory framework on biosafety: Medicinal GMOs for human or veterinary use https://www.biosafety.be/content/eu-regulatory-framework-biosafety-medicinal-gmos-human-or-veterinary-use
- The European Biosafety Association (EBSA) European biosafety / biosecurity legislation https://ebsaweb.eu/european-biosafety-biosecurity-legislation
Dave Li ,MD, PhD, is Principal Consultant; and Anna Baran, MD, is Chief Medical Officer, both of KCR Trial Execution Consulting





Newsletter
Stay current in clinical research with Applied Clinical Trials, providing expert insights, regulatory updates, and practical strategies for successful clinical trial design and execution.












Unifying Industry to Better Understand GCP Guidance
May 7th 2025In this episode of the Applied Clinical Trials Podcast, David Nickerson, head of clinical quality management at EMD Serono; and Arlene Lee, director of product management, data quality & risk management solutions at Medidata, discuss the newest ICH E6(R3) GCP guidelines as well as how TransCelerate and ACRO have partnered to help stakeholders better acclimate to these guidelines.

Managing Side Effects and Dosing in Off-Label GLP-1 Use with Help from Real-World Evidence
July 18th 2025Shipra Patel, global therapeutic area section head, endocrinology, global head, pediatrics, Parexel, explains how real-world data is helping researchers navigate gastrointestinal side effects, dose flexibility, and long-term tolerability in off-label GLP-1 use.


Anselamimab Misses Primary Endpoint in Phase III CARES Trials for AL Amyloidosis
July 17th 2025In the Phase III CARES trials, anselamimab did not meet the primary endpoint for advanced-stage AL amyloidosis, but a prespecified subgroup analysis revealed meaningful improvements in survival and cardiovascular outcomes.


