Consequences of Brexit for Clinical Trials in Europe
Higher costs headline list of new challenges faced by CROs and sponsors.



"Move to EU to avoid Brexit costs, firms told.” This was the news in UK a couple of weeks after the signing of the Trade and Commerce Agreement, as reported by The Guardian.1 The recommendation, directed at all companies of the United Kingdom that export to the European Union (EU), also applies to sponsors of pan-European clinical trials located outside of the new EU27—such as the United States.
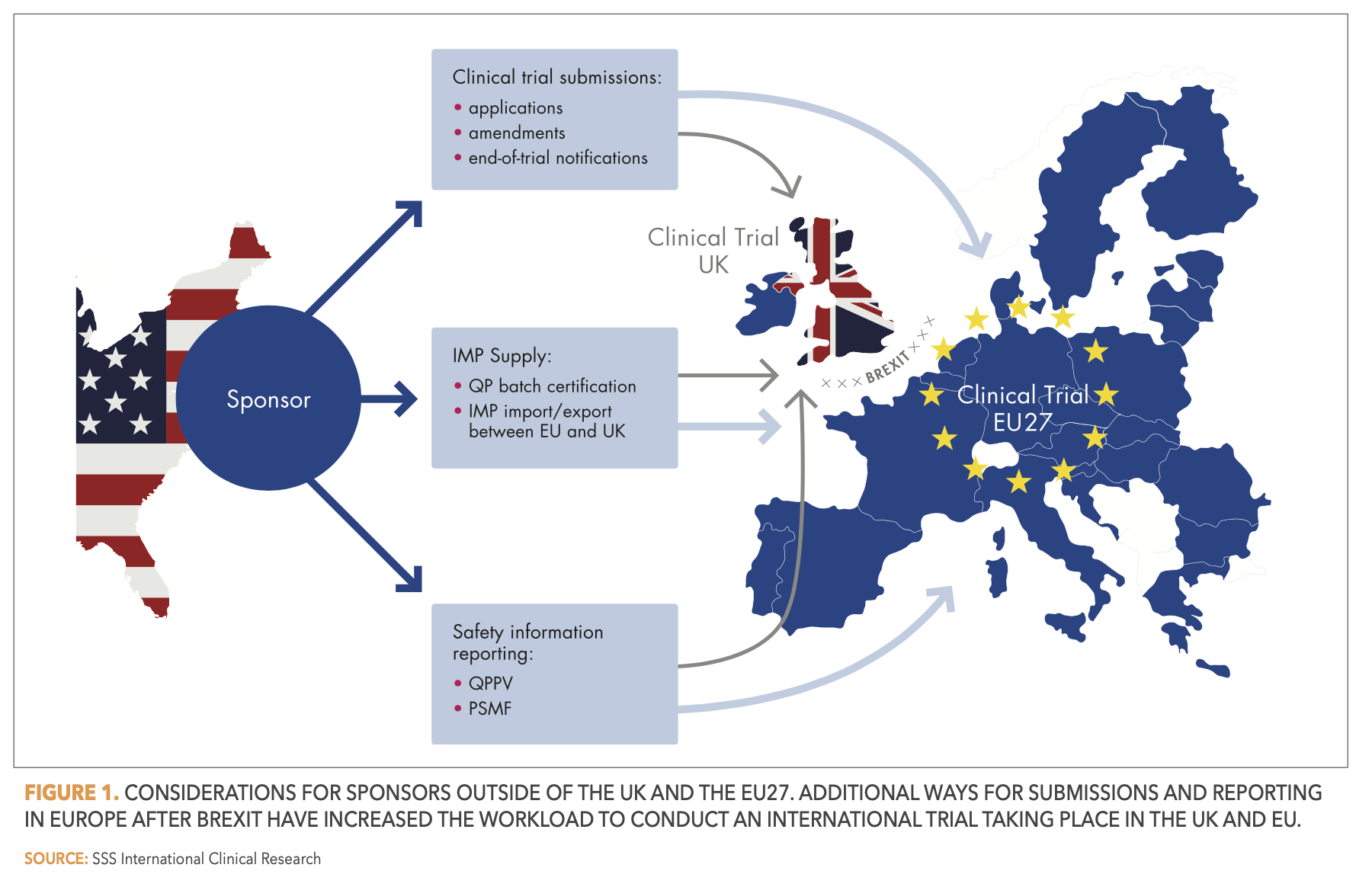
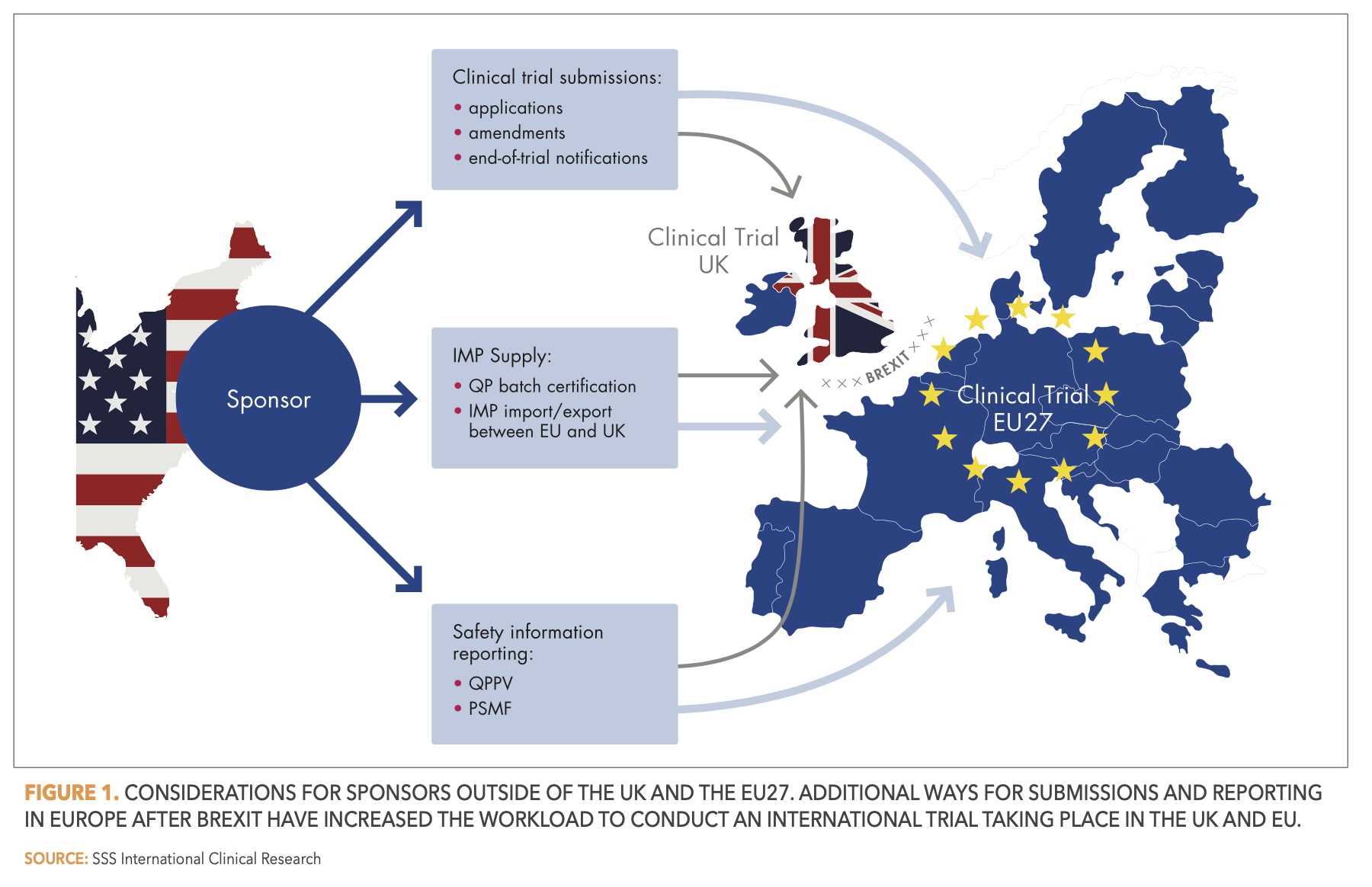
Whether it be the location of the legal representative, the submissions of applications to responsible regulatory agencies, the pharmacovigilance duties to assure the safety of medicines, or the delivery of Investigational Medicinal Products (IMP) to clinical sites: after Brexit, new procedures must be set in place to address all these aspects. There is plenty of guidance information available. Both the European Medicines Agency (EMA) and the British Medicines and Healthcare products Regulatory Agency (MHRA) are continuously publishing and updating relevant information.2,3 In this article, we summarize the most important changes for sponsors located outside of the EU27 that wish to conduct clinical trials of medicinal products in the EU27 and the UK. A visualization of these changes can be found in Figure 1 below.
Sponsor’s location
Starting with the location of the sponsor or legal representative of an EU clinical trial: US pharmaceutical companies have traditionally looked to the United Kingdom for competent and capable partners to conduct clinical trials in Europe owing to the ease of communication that the shared language brings. Unfortunately, solely UK-based CROs can no longer conduct clinical trials in the EU on behalf of sponsors from the US or other non-European locations. EU law demands that the sponsor or legal representative be established in the EU.
In contrast, EU sponsors or CRO partners based in the EU can conduct a clinical trial for medicinal products in both the EU and the UK. In 2019, in its Human Medicines (Amendment etc.) (EU Exit) Regulations,4 the UK introduced an approved countries list that defines where a clinical trial sponsor or their legal representative may be established to be allowed to conduct a clinical trial for medicinal products in the UK. This list initially includes all EU and European Economic Area (EEA) countries and will be reviewed at least every three years by the British government.
Acting as legal representatives for non-EU sponsors—whether from the UK or other parts of the world—CROs must be legally integrated in the EU to be able to conduct clinical trials there. UK-based CROs must establish a presence in the EU if they do not want to be left out.

Applications and pharmacovigilance
Over the lifecycle of a clinical trial, various submissions such as initial applications, substantial amendments, or end of trial notifications must be made. Sponsors or CRO partners of clinical trials for medicines in the EU27 and UK are responsible for these submissions. They can no longer solely use the EU portals—currently still the Common European Submission Portal (CESP) and, from January 31, 2022, onward, the Clinical Trials Information System (CTIS). Instead, they must use the MHRA gateway, as the MHRA is now the standalone medicines and medical devices regulator for the UK.
Similarly, the EU/EEA and the UK are now separate regulatory entities with regards to drug safety. Thus, parallel reporting through the appropriate channels in the EU (EudraVigilance gateway) and UK (MHRA gateway) is necessary.
Accordingly, marketing authorization holders have increased responsibilities in terms of assuring the ongoing safety of marketed products in both regulatory systems. They are required to keep two pharmacovigilance system master files (PSMF) up to date. And although the UK allows the qualified person for pharmacovigilance (QPPV) to be located in the EU or EEA, it is still necessary to define a national contact person for pharmacovigilance in the UK.
CROs representing sponsors in clinical trials taking place in both the EU and the UK, therefore, have a higher workload, as an additional regulatory agency must be addressed.
Time and costs for IMP logistics between EU and UK
An important issue for sponsors is how to ensure continued and unrestricted supply of IMPs to the clinical trial sites. Sponsors of EU and UK clinical trials that import IMPs into Great Britain from outside the UK must reassess their study supply chains after Brexit. They are required to create assurance systems to control whether the IMPs have been duly certified by a qualified person (QP), before release of the IMPs to trial sites. The UK will continue to recognize QP Certification of IMPs from countries on an approved list, which currently includes all EU and EEA countries. As for the abovementioned list of accepted countries for the sponsor’s location, this list of approved countries will also be subject to review at least every three years, introducing a new level of uncertainty in the long-term planning of clinical trials.
For the export of IMPs from Great Britain to the EU, on the other hand, sponsors must organize a new importer and batch releaser, plus a new QP declaration. The only workaround for this is to have the IMP manufactured in Northern Ireland and then ship it to the EU, as it is then not considered an imported IMP under the Northern Ireland Protocol.
This issue will be for CROs coordinating the logistics of the IMP supply to deal with. The simplest scenarios are those in which the IMP is manufactured, packaged, and distributed either in the EU or the UK alone. Sponsors wishing to ship IMPs between the EU27, and the UK should factor in the longer timeframes and additional organizational issues at the very beginning of planning the clinical trial.
Small parenthesis: business as usual for data protection
At the end of June 2021, the European Commission determined that the UK provides an adequate level of data protection and formally adopted adequacy decisions for the UK under the General Data Protection Regulation (GDPR). These allow for the continued unrestricted personal data flow from the EU to the UK. Personal data flows in the opposite direction also remain unaltered, as the British government has similarly decided not to impose additional safeguards. With regard to data protection issues, no adaptation is required by sponsors or CROs after Brexit, regardless of whether they are UK or EU-based.
CTIS simplifies submissions, but not in the UK
As early as in 2014, the EU members adopted the clinical trials regulation (Regulation (EU) No 536/2014), which provides for the central submission of study applications and of documents for European studies. Since the end of 2014, a clinical trial application for multinational studies in the EU can be submitted in accordance with the Voluntary Harmonized Procedure (VHP). In a single submission, a sponsor can simultaneously submit a clinical trial application to the competent authorities in all participating member states of the European Union. But the VHP is only an intermediate solution for a centralized submissions system. On January 31, 2022, the new EU-hosted portal, Clinical Trials Information System (CTIS), will go live. The EMA website states that “CTIS will become the single-entry point for clinical trial application submission, authorization and supervision in the EU, and in the European Economic Area (EEA) countries Iceland, Liechtenstein and Norway.” Through CTIS, sponsors and/or their CRO partners will be able to apply for clinical trial authorization in up to 30 EEA countries with one single application. The same requirements and deadlines will apply to all EU member countries, resulting in a harmonized electronic submission and assessment process for clinical trials conducted in multiple member states. With CTIS, information sharing will be ensured and decision-making processes between and within member states will be expedited. The activation of the new portal will allow Clinical Trials Regulation (Regulation (EU) No 536/2014) to come fully into force at the beginning of 2022—thereby making it easier for trials to be conducted in the EU across multiple member states and making the EU much more attractive for sponsors worldwide. As submissions in the UK will have to be carried out separately via the MHRA, they will not benefit from the simplifications provided by the new CTIS system.
Final practical considerations
In summary, the workload to conduct an international trial taking place in the UK and EU has become heavier. Whereas Brexit has increased the complexity of conducting pan-European clinical trials including the UK, in the EU the Clinical Trials Regulation will make it easier for trials to be conducted across multiple EU member states.
As every clinical project is different, sponsors must carefully select the right European countries in order to bring the clinical trial to successful completion. This success depends on many aspects, including not only the availability of patients in the respective countries but also the availability of dedicated investigators and the treatment praxis.5 The UK is still an important player from a health economics point of view. Nonetheless, sponsors will not automatically include the UK in their clinical program planning and will always evaluate whether the extra effort involved is worth it.
Additionally, regardless of a certain level of economic integration assured by the EU-UK Trade and Cooperation Agreement concluded in late December 2020, insecurities about a stable British partnership remain that may also affect clinical research. Sponsors and CROs should keep a watchful eye over future legislative changes in the United Kingdom. One thing is certain: Planning and successfully performing clinical trials in the EU and UK involves higher costs and more work after Brexit. Sponsors from the US and other non-European countries should take this into account at the earliest possible time point when planning their clinical program for the EU.
References
- Article (British government officials advise companies to expand to EU.)https://www.theguardian.com/politics/2021/jan/23/brexit-hit-firms-advised-government-officials-set-up-shop-in-eu
- https://www.ema.europa.eu/en/about-us/brexit-uk-withdrawal-eu/brexit-related-guidance-companies
- www.gov.uk/mhra.
- The Human Medicines (Amendment etc.) (EU Exit) Regulations 2019, SI 2019/775
- Interview with Lars Behrend from 21.07.2021 by P. Ledesma, Sofpromed, https://www.sofpromed.com/sss-cro-for-clinical-trials-in-germany/
Lars Behrend, PhD, Chief Operating Officer, Ana Cosme, PhD, Clinical Trial Coordinator, and Michael Sigmund, DVM, Chief Executive Office, Founder; all with SSS International Clinical Research









")
















Unifying Industry to Better Understand GCP Guidance
May 7th 2025In this episode of the Applied Clinical Trials Podcast, David Nickerson, head of clinical quality management at EMD Serono; and Arlene Lee, director of product management, data quality & risk management solutions at Medidata, discuss the newest ICH E6(R3) GCP guidelines as well as how TransCelerate and ACRO have partnered to help stakeholders better acclimate to these guidelines.

Funding Cuts Threaten Diversity in Clinical Research
June 27th 2025In this video interview, Kyle McAllister, co-founder, CEO, Trially, discusses how recent federal funding cuts are likely to undermine research focused on underrepresented populations, and why long-term investment in community-based studies is essential to closing persistent health equity gaps.

What Can ClinOps Learn from Pre-Clinical?
August 10th 2021Dr. Hanne Bak, Senior Vice President of Preclinical Manufacturing and Process Development at Regeneron speaks about her role at the company as well as their work with monoclonal antibodies, the regulatory side of manufacturing, and more.


2025 DIA Annual Meeting: Why AI and Automation Are Set to Become the New Normal in Clinical Research
June 20th 2025Peter Ronco, CEO, Emmes, shares his long-term vision for artificial intelligence in clinical research, from making automation routine to improving drug discovery, transforming regulatory oversight, reducing animal testing, and promoting ethical, equitable data use worldwide.

