How to Improve Quality of Study Delivery? Work with Technology-enabled Sites
Practical ways CROs and sponsors can support sites and improve quality.



While a lot has changed in the past five years in clinical research, the most common findings from Food and Drug Administration (FDA) inspection of research sites remain the same.1 Issues such as protocol non-compliance, incomplete case history documentation, and inadequate investigational drug disposition records are the most frequently cited items year after year.
Implementing clinical trial technology at the site level can detect and potentially avoid frequently observed quality issues arising during the trial. Technology systems considered core to site operations include:
- Clinical trial management system (CTMS): A CTMS is responsible for managing all operational aspects of a clinical trial from study start-up to closeout. Critical features include, but are not limited to: study milestone tracking, review/sign-off workflow, calculation of subject visit schedules, financial billing/tracking, and task management.
- eRegulatory Management (eReg) System: An eReg system contains the electronic final copy of site-level essential documents related to study startup and amendments, training documentation, delegation of authority log, and other items needed for audit or inspection. The system also manages subject-level documents such as screening/enrollment log, list of subject identification codes, and signed consent forms. Other items include but are not limited to: protocol deviations/exceptions, safety (AE/SAE) log, and correspondence with the sponsor, contract research organization (CRO), or monitor.
- eSource/Electronic data capture (EDC): eSource and EDC systems capture source data in an electronic format. While there are many similarities between eSource and EDC, eSource will capture every piece of information potentially needed from a standard of care, research, and workflow perspective. EDC is often a subset of eSource because it does not collect protected health information (PHI).
This paper summarizes the most common reasons for findings (often referred to as FDA Form 483 observations) during FDA inspections, and how technology deployed at the clinical trial site can help address these issues.
U.S. FDA inspection findings
The FDA’s Office of Regulatory Affairs conducts inspections and documents any conditions potentially in violation of FDA’s requirements on an FDA Form 483.2 Of particular importance is FDA’s Bioresearch Monitoring (BIMO) program, which provides for the protection of human subjects by assuring the quality and integrity of data submitted to the agency for new product approvals. The FDA summarizes data on inspection findings for each fiscal year tied back to the Code of Federal Regulation referenced in the citation.
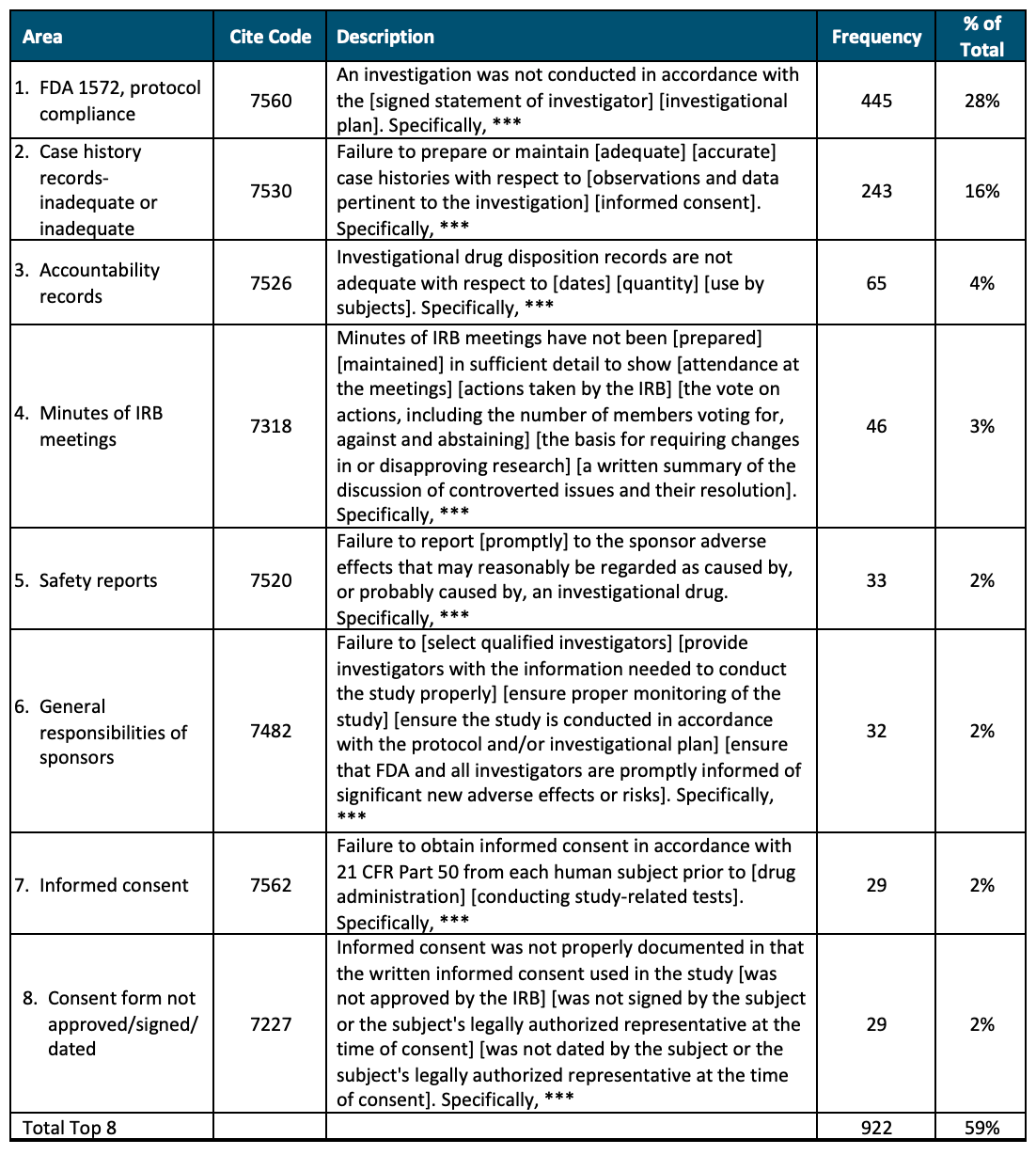
Over the past five fiscal year periods (FY 2018 to FY 2022), there were 763 findings related to bioresearch monitoring. Table 1 below summarizes the top eight reasons for inspection findings. These eight reasons for citation represent 59% of all findings over the five fiscal years; all the other findings represent less than 1.5% of the total related to inspections.


Source: Advarra analysis of FDA Inspection Observation Datasets, FY 2018-FY 2022. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-observations")
Table 1. Five-year U.S. FDA Inspection Findings (FY 2018 to FY 2022)
Source: Advarra analysis of FDA Inspection Observation Datasets, FY 2018-FY 2022. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-observations
Improving study delivery quality via site technology
As described above, high volume clinical trial sites and site networks generally invest in three types of technology: CTMS, eReg binders, and eSource/EDC. In addition to standardizing the research process at the site, each of these technologies has the potential to address the reasons for common FDA Form 483 inspection findings related to biomedical research monitoring.
CTMS
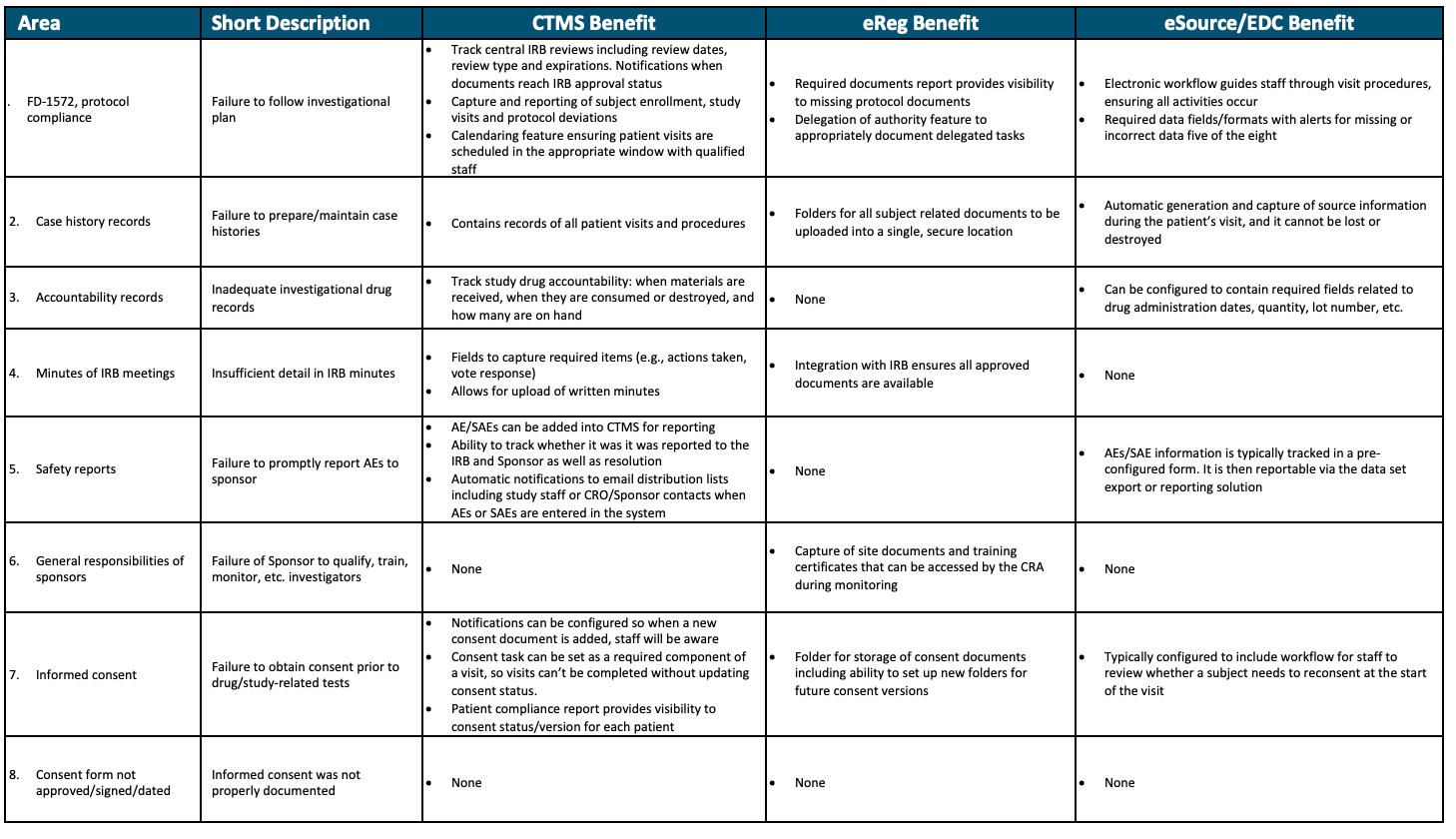
Sponsor benefits from CTMS usage are summarized in Table 2. Adopting a site CTMS can prevent and/or detect six of the eight most common observations during an FDA inspection. The two common observation items not covered by CTMS would generally be the responsibility of the study sponsor, as opposed to the site. Outside of these eight common compliance issues, site-level CTMS also can also improve patient recruitment and retention, support financial compliance and insurance versus study billing (i.e., coverage analysis), and guide study startup staffing and timelines.
eReg System
As seen in Table 2, an eReg system provides benefit in five of the eight most common compliance issues found during FDA inspection. One of the three items not covered by eReg is typically a sponsor responsibility. In addition to the benefits displayed in Table 2, an eReg system also helps decrease study startup timelines by leveraging documents across trials, as well as facilitating capture of signature for eConsent.
eSource/EDC
It is generally accepted that electronic capture of data via eSource offers benefits over paper, improving timeliness and quality of data. But it may not be as well-known that eSource and EDC systems decrease risk associated with five of the eight common findings displayed in Table 2. Similar to CTMS, two of the three areas not covered are typically sponsor responsibilities. Also seen in Table 2, the benefits from CTMS, eReg, and eSource/EDC differ, providing a variety of benefits for the large proportion of sites who use all solutions in combination.


Table 2. Quality Benefit from Site CTMS, eReg, and eSource/EDC Platforms
How can clinical research sponsors and CROs help support use of site technology?
Sites invest in technology at the enterprise level for use across all studies and sponsors, including industry, government, and investigator-initiated trials (IITs). Sponsors and CROs wanting to support sites in using their own technology to drive more compliant and higher quality study delivery can help in the five ways listed below.
- Pre-qualification of software tools: Today, evaluation of a site’s technology occurs at the individual site level despite the fact that CTMS, eReg, and eSource/EDC technology is offered by a small number of providers. Instead of visiting each site, sponsors have the potential to decrease site burden as well as improve their own efficiency and cycle times by centrally reviewing and qualifying technology.
- Schedule of events/calendar builds in CTMS: One of the steps in setting up a CTMS system for a new study is to build the protocol schedule of events into the calendar function. While sponsors and CROs should consider adding a line item for reimbursement for this system setup task in the site’s budget, perhaps a more time saving approach would be to ask individual technology vendors to perform the configuration centrally and distribute the module to all their site technology customers working on the trial. The information can also be provided in a spreadsheet as a visit calculator tool for technology-naïve sites.
- Budget setup in CTMS: Similar to calendar builds, there is also an opportunity to insert a line in the site budget template for budget setup or to set up the budget module centrally and distribute the tool to all site technology customers. This avoids setup fees at the site level and saves the site time in study startup.
- Regulatory-compliant coverage analysis (U.S. only): This activity entails classifying which study procedures can be billed to insurance (e.g., Medicare). While coverage analysis is valuable to all sites, building the information into the CTMS provides added value in automated financial and billing compliance (e.g., insurance vs. study budget). Savings from this support activity can be substantial. A recent AACI white paper published in April 2023 suggests the potential to save five to 10 business days in budget discussions during startup from sponsor-provided coverage analysis. Specifically, the paper states: “…the goal for consensus budget is 25 business days. This can be further improved (our estimate is 5-10 days less) if the sponsor provides the site a study-specific National Coverage Analysis (NCA) that will detail all procedures that are research (R) and not standard of care (SOC).”3
- Enable integrations with existing site technology (i.e., bring your own technology [BYOT]): When it comes to technology, not all sites have the same needs. For example, there are differences in finance and billing needs between sites providing patient care vs. those solely focused on clinical research; there are differences in reporting requirements for sites (e.g., NCI-designated centers, NIH grants), and site networks/SMOs have a need for central permissions and reporting across all participating sites. In addition, switching costs are high for sites who have already implemented a CTMS, eReg, and/or eSource/EDC system due to integrations, custom processes and reporting, and time and resources for change management. As a result, a single solution meeting both site and sponsor/CRO needs is unlikely to be feasible. Instead, sponsors and CROs should increasingly design their study delivery processes and technology to integrate with the site’s own tools to decrease time and cost of duplicate activities. Integration of a site’s eReg system into sponsor’s or CRO’s document exchange technology and TMF are great examples of integrations delivering better cycle times and quality at a lower cost to the site.
Providing support through the five mechanisms listed above offers a “win-win” situation for sites, sponsors, and CROs: a decrease in site burden related to technology and an improvement in sponsor and CRO efficiency, quality, and compliance in study delivery.
Conclusion
The reasons for quality findings related to clinical trials are well documented, and many experienced sites have chosen to purchase technology to decrease risk associated with protocol compliance, inadequate case histories, and accountability records (i.e., drug disposition), among other issues. What is not well known, is how this technology benefits sponsors and CROs. In fact, the majority of sponsors and CROs who oversee clinical trials are not at all familiar with site technology and its value.
Press releases, websites, and participation of sponsors and CROs in clinical trial workforce associations such as the Association of Clinical Research Professionals (ACRP) all point to a strong desire to decrease site burden, but there's not always a clear, actionable path for sponsors and CROs to address the problem. The five suggestions listed above—pre-qualifying software tools; calendar builds, budget setup, Medicare coverage analysis in CTMS, and offering BYOT—are practical ways sponsors and CROs can support sites and ultimately help themselves through faster startup and higher quality study delivery. Recognizing sites are already struggling to achieve workforce levels to keep pace with industry clinical trial volume, anything sponsors and CROs can do to decrease site burden will likely have a positive impact on the clinical trial industry, well beyond an individual trial.
Elisa Cascade, chief product officer, and Kate Yawman, director, product management; both with Advarra
References
- https://www.aaci-cancer.org/Files/Admin/2023-April-Trial-Activation-White-Paper.pdf
- Kingsley, Jeff: Sites Need Technology, Infrastructure Investments to Improve Clinical Trials. Clinical Leader November 29, 2018. https://www.clinicalleader.com/doc/sites-need-technology-infrastructure-investments-to-improve-clinical-trials-0001
- U.S. Food and Drug Administration, Inspection Observations: November 21, 2022: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-observations.












Improving Relationships and Diversifying the Site Selection Process
April 17th 2025In this episode of the Applied Clinical Trials Podcast, Liz Beatty, co-founder and chief strategy officer, Inato, discusses a number of topics around site engagement including community-based sites, the role of technology in improving site/sponsor relationships, how increased operational costs are impacting the industry, and more.




