Analysis: FDA’s COVID-19 Accelerated Pathways
Lessons learned from FDA’s current process and proposed alternative review strategies.



At the October 2021 DIA conference on Digital Technologies in Clinical Trials, Pfizer reported the following timeline for the Pfizer-BioNTech COVID-19 vaccine
clinical program:
On May 4, 2020, the Phase I study was initiated, followed by initiation of the Phase II/III program on July 27, 2020. Positive interim results were reported on November 18, 2020 followed by the Emergency Use Authorization (EUA) submission request on November 20, 2020. The first Food and Drug Administration (FDA) EUA clearance occurred on December 2, 2020. The program, which evaluated over 46,000 subjects at approximately 150 clinical trials sites in United States, Germany, Turkey, South Africa, Brazil and Argentina, took just 8 months from first patient first visit (FPFV) to FDA clearance.
This major achievement by Pfizer, as well as the rapid development of products to prevent and treat the novel coronavirus disease 2019 (COVID-19) by others in the pharmaceutical industry and academic centers, are to be commended. Similarly, the FDA must also be commended on the speed that products have received EUA and full marketing approvals during the COVID-19 pandemic.
While clinical trials are costly and time consuming, they are necessary to assure that safe and effective products reach patients. As it is well known, the steps of drug development to approval involve multiple factors that could be improved in order to accelerate the process. The processes, resources and strategies to maximize efficiencies need to be revisited in order to continue exploring innovative study designs and methodologies that could result in reducing timelines and costs to product approval. On this topic, a recent article addresses the value of randomized clinical trials as well as the value of real world evidence together with novel approaches to statistical methodologies.1 Another recent article addresses the suitability of registries for embedding clinical trials.2
Going forward, efforts need to be magnified to encourage biotech and pharmaceutical companies to continue promoting solutions to benefit of patients who are expecting solutions for their unmet medical needs. With this premise in mind, the current article first describes recent approaches to accelerate the marketing approval process, followed by a proposition of alternative strategies to accelerate the regulatory review approval process once it is concluded that an investigational product has passed the initial threshold of demonstrating safety and effectiveness in one or more clinical trials.
Current FDA programs to accelerate access
In order to accelerate the product approval process, four regulatory programs currently exist to reduce development and review times for products that address unmet medical needs for the treatment of serious or life-threatening conditions. In May 2014, FDA issued a Final Guidance for Industry entitled, “Expedited Programs for Serious Conditions–Drugs and Biologics” which addresses fast track designation, breakthrough therapy designation, priority review designation and accelerated approval.3
In addition to this guidance document, under section 564 of the FD&C Act, 21 U.S.C. 360bbb-3, in a situation where the Secretary of Health and Human Services (HHS) issues a declaration of emergency or threat justifying authorization of emergency use for a product caused by chemical, biological, radiological or nuclear (CBRN) agents, as well as an infectious disease, the Commissioner of the FDA may authorize an EUA of an unapproved product or an unapproved use of an approved product. In 2004, the EUA authority was incorporated in the Project BioShield Act of 2004, which amended the FD&C Act. In January 2017, the “Emergency Use Authorization of Medical Products and Related Authorities” guidance was finalized.4
Fast Track designation
Section 112 of the Food and Drug Administration Modernization Act of 1997 (FDAMA), entitled “Expediting Study and Approval of Fast Track Drugs,” mandates the facilitation of the development and processes to expedite review of therapeutics intended to treat serious or life-threatening conditions presenting with unmet medical needs. Fast Track designation, which also allows for the possibility of a “rolling review” of New Drug Applications (NDA) and Biologics License Applications (BLA), can be requested early in the development process at the time of Investigational New Drug (IND) filing or completion of Phase I studies. When a product receives a Fast Track designation, there is a strong emphasis on the importance of continuous communication with FDA so that questions and issues can be resolved quickly. Examples of diseases/therapeutic areas considered to be serious conditions include AIDS, Alzheimer’s disease, heart failure, cancer, epilepsy, depression, and diabetes, among others.
Breakthrough Therapy designation
In contrast to Fast Track designation, a Breakthrough Therapy designation is assigned to an investigational product that is intended alone or in combination with one or more other products to treat a serious or life-threatening condition, and where there is preliminary clinical evidence that indicates that the investigation product may demonstrate substantial clinical benefit over existing therapies. Breakthrough Therapy designation requests usually occur at the end of Phase II. A drug that receives Breakthrough Therapy designation is eligible for all of the features of Fast Track designation.
Accelerated Approval
The Accelerated Approval Program supports the concept that a treatment may be conditionally approved based on a validated surrogate biomarker or an intermediate clinical endpoint that strongly support the possibility of clinical benefit. Accelerated Approval agreements with FDA occurs close to the NDA or BLA submission. The regulation also requires that upon receiving Accelerated Approval, sponsors must perform a confirmatory study to demonstrate clinical benefit. If data from the confirmatory study does not demonstrate a clinical benefit, the product will be withdrawn from the market for use in that indication.

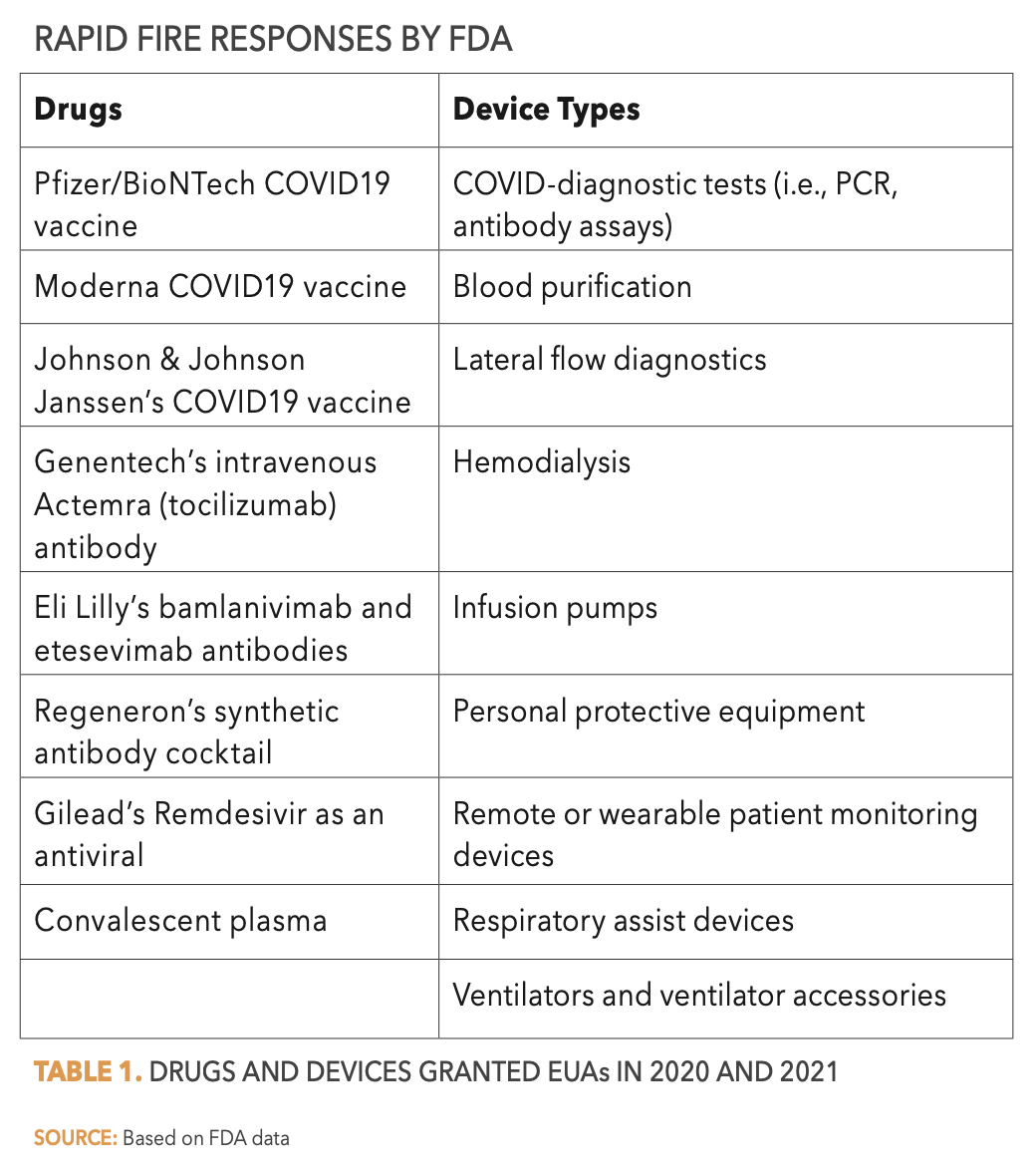
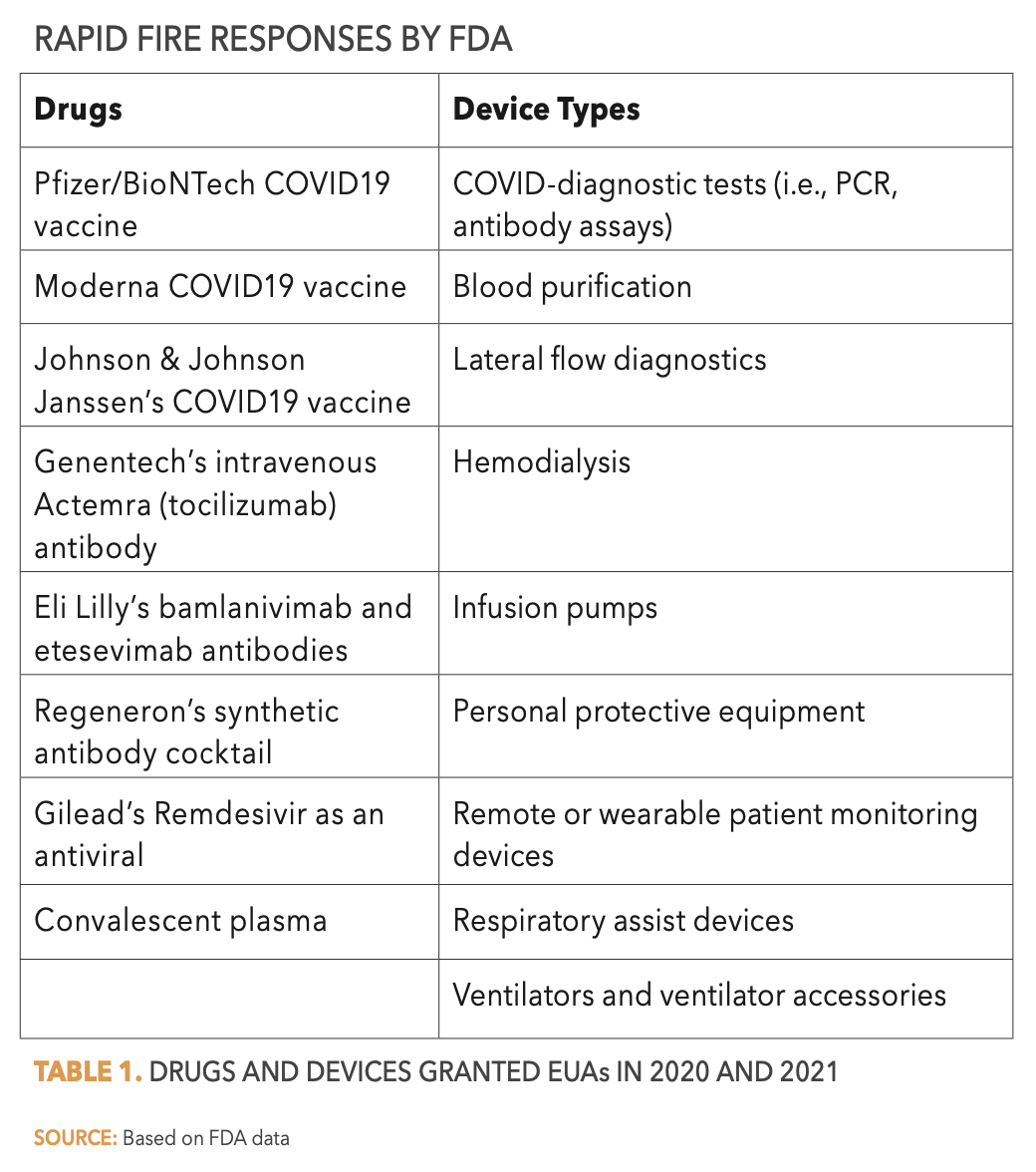
Priority Review designation
Under the 1992 Prescription Drug User Act (PDUFA), FDA committed to specific goals for improving drug review times. As a result, a two-tiered system of review times, Standard Review and Priority Review was created. A Priority Review designation means that the goal is for the FDA to act on a marketing application within 6 months of receipt, rather than a Standard Review of 10 months, as long as the investigational product can be shown to provide significant advantages such as:
- Evidence of increased treatment effectiveness
- Prevention or diagnosis of a condition
- Elimination or substantial reduction of a treatment-limiting adverse drug reactions
- Documented enhancement of patient
compliance - Evidence of safety and effectiveness in a novel subpopulation of a disease
FDA informs the applicant the status of a Priority Review designation within 60 days of the receipt of the original BLA, NDA, or efficacy supplement.
Current FDA approaches to accelerate clearance of medical devices
In 1997, in order to improve the efficiency and timeliness of the 510(k) process of certain low-to-moderate risk new medical devices, the Accredited Persons Program was included in FDAMA. In 2018, a slide deck was published by FDA that describes how it is updating its 510(k) Third Party (3P) Review Program in order to avoid the routine re-review of 510(k) submissions already reviewed by a 3P Review Organization.5 As a follow-up, a March 12, 2020 final guidance document was issued entitled “510(k) Third Party Review Program Guidance for Industry, Food and Drug Administration Staff, and Third Party Review Organizations.”6 This guidance document provides medical device manufacturers with a voluntary process where FDA-authorized Third Party Review Organizations (3P510k Review Organizations) are permitted to evaluate and recommend approval or rejection of new devices. The guidance also delineates how decisions are made if certain medical devices are eligible for review under the Third Party Review Program. This new authority had been codified in the FDA Reauthorization Act of 2017 (FDARA) and allows discretion as to which devices are eligible for third party review. Approximately 50% of 510(k) applications now fall under this program.
FDA post COVID-19
The following section addresses proposed changes to existing FDA rules and regulations using a parallel format of the four phases of clinical trials during drug development. However, before moving forward, the are questions of special interest that should be addressed:
- Under whose authority would it be to propose and implement these changes?
- Would these proposed changes all fall within the jurisdiction of FDA rules and regulations, or would some require legislative action such as modifying the FD&C Act?
Phase 1 - Premarketing
Based on lessons learned from the product approval process during COVID-19, and as previously published over 20 years ago, it is critical that sponsors continue to interact with FDA to discuss their development plans.7 In addition, FDA should expand the scope of exempt studies falling under IND and Investigational Device Exemption (IDE) regulations, and thus allow sponsors, with FDA concurrence, to proceed to clinical trials without FDA clearance of an IND or IDE. However, in order to control this process, each sponsor would be certified through an independent audit, that it complies with all relevant FDA rules and regulations, and training procedures are in place to guarantee that experienced staff manage all aspects of Good Laboratory Practices (GLP), and Good Manufacturing Practices (GMP) and Good Clinical Practices (GCP). The auditors will be certified by FDA based on their curriculum vitae and experience, and there will be an agreed upon fee across the industry, which will be paid by the company being audited. Each company will do yearly internal audits to assure continued compliance, and at the time of a marketing application, this process will be subject to regulatory review during preapproval inspections.
In addition, as is current practice, a clinical trial cannot be initiated without: 1) review and approval of the protocol and informed consent by an IRB or Ethics Committee; 2) review and signoff by an independent study Principal Investigator; and 3) review and signoff by each clinical site. Some academic organizations also have separate protocol review committees that must approve all new protocols.
From the data analysis perspective, the regulatory guidance ICH-E9 R1 is a key instrument to ensure more robustness and consistency during protocol development and statistical analysis planning. In order to minimize bias and increase the precision around the interpretation of the estimates associated with the clinical trial, the guidance forces trialists to provide more details about how the research questions, objectives, endpoints, and statistical analysis should be codified.8
In terms of study designs, the discrepancies between the requirements and magnitude of the thresholds to demonstrate clinically significant safety differences, in most cases, is usually higher than the expectations for the efficacy profile. Thus, to have confidence that a study design is adequate to establish that an investigational product shows success both in terms of efficacy and safety in relationship with a comparator arm, the following issues must be addressed in all applicable phases of clinical development:
- Clearly define the research question and hypothesis
- Definition of the estimand and its attributes for the evaluation of all endpoints, including:
- The population of interest
- Treatment conditions
- The endpoints and variables to be evaluated, and the validated instruments to be used for each case
- Intercurrent events (ICE) that could impact the evaluation and interpretation of all endpoints, and the approaches to be used for each ICE
- Population level summary for the evaluation of endpoints including the analysis dataset that will be used for each case
- Sensitivity and supplemental analyses, for example:
- Using the per protocol population or completers
- Considering other missing data mechanisms different to the assumptions to the one used in the evaluation of the primary endpoint
- Tipping point analysis under different scenarios
- Supplemental analysis using different statistical models/methods
- Missing data strategies
- Testing strategies to control the familywise type I error
- Assessments to capture the data associated with all endpoints using validated instruments
- Description of the evaluation of the safety profile of drugs and devices including:
- Adverse Events (AEs)
- Treatment Emergent Adverse Events (TEAEs)
- Serious Adverse Events (SAEs)
- Treatment Emergent Serious Adverse Events (TESAEs)
- Adverse Events of Special Interest (AESI)
- Serious Unexpected Serious Adverse Reactions (SUSARs)
Phase 2 - Product approval
A general framework currently exists when FDA requests product approval recommendations from Scientific Advisory Boards for drugs and biologics and Third Party Review Organizations for devices. Thus, in order to expedite the review of marketing applications and reduce the time for a final marketing decision, in lieu of a detailed FDA review of all marketing applications, it is proposed that a framework be established for drugs, biologics and high risk new medical devices such that external expert committees, comprised of individuals certified by FDA, also be able to review the marketing applications and make product approval or rejection recommendations to the FDA review divisions responsible for final decision-making. However, in the event there are disagreements or basic questions that need to be resolved during the committee review, a process must be put in place for FDA, the expert review committee and sponsor to rapidly reconcile any and all issues.
For drugs and devices approved outside of the United States, the same processes should be followed by the expert committees, while taking under consideration any significant differences in regional differences in study designs and/or the population(s) studied.
Phase 3 - Post marketing safety monitoring
Currently, companies with FDA-regulated marketed products are required to track and report post marketing safety events either as expedited reports or as part of required annual reporting. Examples of the sources of these safety events include ongoing clinical trials, direct reports to companies of adverse events, data from electronic medical records, foreign data reports, the literature and data from patient registries and professional societies. Patient advocacy groups like the Cystic Fibrosis Foundation and professional societies like the Society for Thoracic Surgeons maintain patient registries.9,10 The Cystic Fibrosis Foundation registry tracks adverse events for all marketed and off label use of drugs for the treatment of patients with cystic fibrosis. In 1989, the Society for Thoracic Surgeons established the STS National Database to support the quality of patient care and to track safety events occurring during surgical procedures performed by their members. While some patient registries are being mandated by FDA to be used for safety monitoring, the use of patient registries should be expanded especially in rare diseases and in special patient population where there are active registries. However, if registries are mandated as a source of safety monitoring, the cost associated with this requirement should be reasonable and transparent so as remove any burdens, especially on smaller companies.
Phase 4A - The Sentinel System
In the Food and Drug Administration Amendments Act (FDAAA; H.R. 3580) of 2007, Congress mandated that FDA track safety signals of marketed products. Subsequently, the Sentinel System was established in 2009 that uses electronic health data to assess post‐market medical product safety.11,12 The full Sentinel System was officially launched in February 2016. Through Sentinel, FDA assesses the safety of approved medical products including drugs, biologics, and medical devices. Sentinel’s data are based primarily on administrative claims and electronic health records (EHRs). The core Data Partner organizations include national and regional health plans partners, integrated delivery systems, and Medicare‐fee‐for service data. Sentinel partnerships also include EHR‐based organizations and networks, including CA Healthcare, the National Patient‐Centered Clinical Research Network (PCORnet), and several EHR data aggregators (TriNetX, Veradigm, IBM Explorys) who contribute data on a case‐by‐case basis.
There are currently over 70 million patients actively providing data to Sentinel. However, since there is a reported lag of 6-9 months from the date of health care and the data are only refreshed quarterly,13 we are pleased that FDA’s FY 2022 budget includes $82.9 million for an agency-wide data modernization and enhanced technologies initiative that supports medical product safety programs. These investments will modernize FDA’s data infrastructure by using current technology innovations that will allow FDA to more efficiently to gather data; identify, analyze, and respond to potential problems more quickly; and further improve review times for medical products.14
Phase 4B – National pharmacy chains
Data from large national pharmacy chains can also be used to rapidly capture and report adverse events immediately upon product launch. For example, on May 20, 2021 CVS announced that their “CVS Health Clinical Trial Services is working with key stakeholders in the biopharmaceutical industry and across the clinical trial ecosystem to design and deliver innovative approaches to research and real-world evidence generation.”15
Conclusion
While COVID-19 and previous pandemics have brought terrible devastation throughout the planet, government and societal reactions to COVID-19, for the most part, have demonstrated that it is possible to develop and release vaccines and other therapeutics in record times, and that humans have the ability and capability to collaborate for the common good. Regulators have also demonstrated that it is possible to accelerate the approval of much needed therapeutics in novel ways, and that if the proper resources are allocated both within and outside of governments, it becomes a win-win for citizens today and for all future generations.
References
- Edwards, SJL, Bock, T., Palm, U., et al. The Case for Methodological Pluralism in Medical Science, The American Journal of Bioethics, 20:9,
39-41. 2020 - Mikita, S., Mitchel, J., Gatto, NM, et al. Determining the Suitability of Registries for Embedding Clinical Trials in the United States: A Project of the Clinical Trials Transformation Initiative. Therapeutic Innovation & Regulatory Science. 2020
- FDA: Guidance for Industry Expedited Programs for Serious Conditions – Drugs and Biologics. 2014
- FDA: Emergency Use Authorization of Medical Products and Related Authorities. 2017
- FDA: Eliminating Routine FDA Re-Review of Third Party 510(k) Reviews. 2018
- FDA: 510(k) Third Party Review Program Final Guidance for Industry, Food and Drug Administration Staff, and Third Party Review Organizations. 2020
- Mitchel, J. FDA Relations During Drug Development. Dialogues in Clinical Neuroscience. 2(3): 213–217. 2000
- ICH: E9(R1) Statistical Principles for Clinical Trials: Addendum: Estimands and Sensitivity Analysis in Clinical Trials. 2021
- Cystic Fibrosis Registry Patient Registry | CF Foundation. 2019
- Society for Thoracic Surgeons: STS National Database. 2021
- How Sentinel Gets Its Data - Sentinel Initiative. 2019
- Cocoros, NM, Fuller, CC, Adimadhyam, S., et al. A COVID‐19‐Ready Public Health Surveillance System: The Food and Drug Administration’s Sentinel System. Pharmacoepidemiology and Drug Safety. 30(7): 827–837. 2021
- Martinez, AI. Monitoring Medication Use During the COVID-19 Pandemic in the Sentinel System: The Case of Anticoagulation for Thrombosis. 2021
- FDA Press Release: FDA’s Budget: Data Modernization and Enhanced Technologies. 2021
- CVS Health introduces Clinical Trial Services | CVS Health. 2021
Jules T. Mitchel, MBA, PhD, President, CEO, THI Pharma Services Inc., Luis A. Rojas, PhD, President and CEO, InCSD, and Jonathan S. Helfgott, MS, Faculty, Program Coordinator, Senior Lecturer, MS in Regulatory Science & Food Safety Regulation Programs, Johns Hopkins University









")
















Unifying Industry to Better Understand GCP Guidance
May 7th 2025In this episode of the Applied Clinical Trials Podcast, David Nickerson, head of clinical quality management at EMD Serono; and Arlene Lee, director of product management, data quality & risk management solutions at Medidata, discuss the newest ICH E6(R3) GCP guidelines as well as how TransCelerate and ACRO have partnered to help stakeholders better acclimate to these guidelines.


FDA to Launch National Priority Voucher Program to Speed Drug Reviews for Critical Therapies
June 18th 2025Under the new initiative, companies may receive a voucher enabling FDA review to be shortened from the standard 10–12 months to just 1–2 months following final application submission if the drug addresses US national health priorities.



