News
Advertisement
Company News Release
Aspire IRB announces record growth for the third consecutive year, application for accreditation and a newly established web portal
Advertisement
Company News Release
Covance Inc. (NYSE:CVD), a provider of drug development services, today announced that its clinical pharmacology services were recognized for outstanding customer service by Eli Lilly and Company.
Company News Release
Covance Inc. (NYSE:CVD), a provider of drug development services, today announced that its central laboratory services received Merck Co., Inc.'s Outstanding Strategic Collaboration Award.
Company News Release
Metroplex Clinical Research Center (MCRC), a multi-specialty clinical research site in Dallas, has enjoyed a 250 percent increase in business since site executives announced plans to reclaim its assets from Radiant Research in March 2007.
Company News Release
TUV SUD has completed the acquisition of all shares of MSOURCE Medical Development to create a new T?V S?D Life Science business unit.
Advertisement
Company News Release
CEO Chairman Jim Walker of Octagon Research Solutions, Inc., a provider of software and services to the life sciences industry, announced today that a new Electronic Data Capture (EDC) Blog entitled, ?EDC Connections? Discussion Forum is now available on the Octagon Research Solutions, Inc. website.
Company News Release
Pharmatech, Inc., a Research Management Organization (RMO), has contracted with five leading pharmaceutical companies to provide research and site management services for their clinical trials using the Just-in-Time (JIT) enrollment strategy.
Company News Release
Velos, Inc., announced today its plans to implement Phase II of its Strategic Plan ? an integration with third party systems and systems-based collaboration across research sites and sponsors.
Company News Release
Abingdon Life Sciences, Inc. is a Drug, Device, Diagnostic Development Management Organization (DDMO), focused on providing drug and device companies with superior services based on an integrated strategic development model.
Advertisement
Company News Release
Pharsight Corporation (Nasdaq:PHST), a provider of software, strategic consulting, and regulatory services for optimizing clinical drug development, and CRI Worldwide, a provider of clinical research services to the global pharmaceutical and biotech industries, announced that they have formed an alliance to offer combined services.
Company News Release
Health Market Science, Inc., a leading source of healthcare provider data and analytics solutions in the United States, announced the launch of Clinical Investigators, their latest innovation for clinical site recruitment.
Company News Release
Praxis, a company specializing in centralized patient recruitment for clinical research studies, was recently awarded Best of Show as well as two Gold International Awards of Excellence (IN-AWE Awards) by the Healthcare Communication & Marketing Association (HCMA)
Company News Release
ProTrials Research, Inc.TM, a leader in the clinical research organization industry, announced the Silicon Valley/San Jose Business Journal has chosen it as one of the area?s Top 50 Women-Owned Businesses for 2008. ProTrials placed seventh on the list that ranks area public and private women-owned companies according to revenue.
ACT's eNewsletter, Lab Views, brings you news specific to the laboratory and its uses in clinical trials including deals, alliances, business developments, people news, and events.
Company News Release
Thermo Fisher Scientific Inc., announced that Veeda Clinical Research has selected its Thermo Scientific Watson LIMS to automate its laboratory processes in its Bioanalytical Research facilies in Ahmedabad, India and Oxford, UK
Company News Release
FDA changes "approvable" and "non-approvable" letters to "complete response" letter at the end of the review period.
Company News Release
The Association of Clinical Research Organizations (ACRO) recently announced the election of Dr. Derek Winstanly as Chair-Elect of the association.
Company News Release
The URMC will collaborate with the FDA to develop a national repository of data that will aid academic and industry researchers studying the electrical activity of the heart.
Company News Release
Almac Clinical Technologies announced today that its Testing Department has completed the ISTQB Software certification.
Company News Release
ClinPhone, a Clinical Technology Organization (CTO) and Cytel Inc., statistical experts in adaptive clinical trials, announced a partnership that unites expertise and clinical technologies
Applied Clinical Trials
A recently released report details the current status of the clinical trials market in Russia.

Applied Clinical Trials
Industry news focusing on the people and organizations who work in the clinical trials profession.

Applied Clinical Trials
PharmaPros Takes Electronic Data Lifecycle Management to a New Level with Dataflow Manager.

Applied Clinical Trials
Online survey allows clinical research professionals to share their experiences with the European Clinical Trials Directive.
Company News Release
The Center for Information and Study on Clinical Research Participation (CISCRP) recognized PharmaNet as this year?s award recipient for ?Supporting the Development of Educational Materials.?
Applied Clinical Trials
Lawke Links, Cordium Links new joint venture with a Chinese organization, will bring a full range of ECG core lab services to China.
Applied Clinical Trials
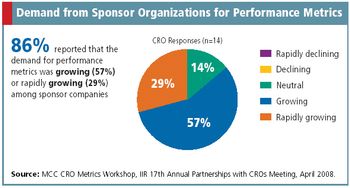
Metrics Champion Consortium standardizes CRO performance metrics across the board.
Company News Release
Symbio LLC, a full service contract research organization, has announced the opening of a new Phase I Clinical Unit in Michigan City, Indiana.
Advertisement
Advertisement
Trending on Applied Clinical Trials Online
1
Real-Time Trials Need More Than AI: Why Operational Trial Infrastructure Matters
2
FDA Makes Case for Overhauling Early Clinical Development to Reclaim US Leadership
3
ACT Brief: Commercial Strategy Shaping Early Clinical Development, Real-Time Trials Require Operational Infrastructure, and AI Site Engagement Gap
4
ACT Brief: Funding Gaps and Indication Decisions in Early-Stage Programs, Recruitment Funnel Leak Diagnosis, and Mega-Merger Speculation Denial
5