



Applied Clinical Trials
EU Council supports pediatric medicines proposal, but adds some changes.

Applied Clinical Trials
EU Council supports pediatric medicines proposal, but adds some changes.

Applied Clinical Trials
The much anticipated list features initiatives in biomarkers and research productivity.

Applied Clinical Trials
Online trial registries alone will not succeed at rebuilding public confidence.
Applied Clinical Trials
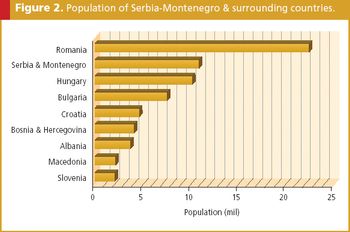
Out of the remnants of Yugoslavia, Serbia has reasserted itself on the European pharmaceutical stage.

Applied Clinical Trials
Biomarkers take center stage in Europe, where their critical role is under examination.

Applied Clinical Trials
More consultations with sponsors promise to drive drug development, but also impose a growing burden on FDA.

Applied Clinical Trials
Personal integrity is what we need to insist on as the finality of good clinical practice.
Applied Clinical Trials
The impact of the EU Clinical Trials Directive on current clinical research practices

Applied Clinical Trials
A system of checks and examinations that helps ensure the quality of clinical trials.

Applied Clinical Trials
FDA seeks to reduce clinical research failures and make drug labeling more useful.

Applied Clinical Trials
When negative results arise, novel analysis packages help speed the decision to move forward or pull the plug on a new drug.

Applied Clinical Trials
As patents expire, clinical trials will be caught in the middle between copyists and original innovators.

Applied Clinical Trials
Today's technology makes significant improvements in trial safety systems possible.

Applied Clinical Trials
Drug safety and R&D issues will play a prominent role in user fee program revisions.

Applied Clinical Trials
Changes prompt new oversight approaches by FDA and research institutions.

Applied Clinical Trials
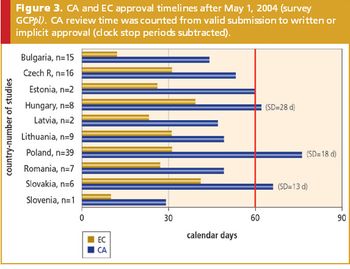
As some EU countries struggle with the Directive's regulatory demands, emerging markets such as China, India, and Russia continue to bolster their clinical research and drug development programs.

Applied Clinical Trials
GRPs are not another set of guidelines or "additional hurdles" prepared by authorities.

Applied Clinical Trials
New paper from the European Medicines Agency explores future vaccine guideline.

Applied Clinical Trials
While veteran officials keep the agency running, changes continue at FDA's new drug review and safety oversight offices.
Applied Clinical Trials
A comprehensive listing of U.S. departments and offices that includes the telephone numbers of directors, commissioners, and advisors.

Applied Clinical Trials
Three recent initiatives address the reputation of the health care industry in the EU.
Applied Clinical Trials
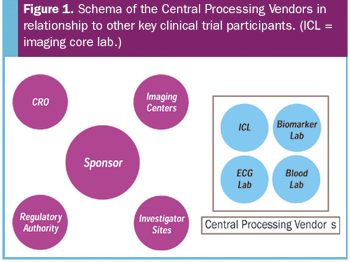
Technological advances have lead to an increasingly important role for centralized imaging laboratories.
Applied Clinical Trials
Implementing ICH E14 to define cardiac safety in new drug development

Applied Clinical Trials
Janssen Pharmaceutica and Medidata Solutions join forces to institute eSourcing.