While not setting any records for the rapid approval of new drugs in 2019, FDA did speed a number of important new therapies to patients, writes Jill Wechsler.
FDA
Latest News
Advertisement
Advertisement
Advertisement
Applied Clinical Trials
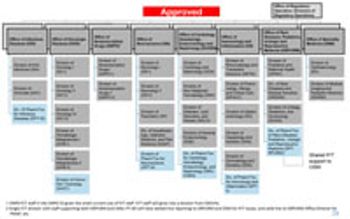
FDA’s Office of New Drugs restricting aims to improve scientific exchange and information sharing among review professionals.

Applied Clinical Trials
Examining the unique aspects involved in preparing and submitting marketing applications for proposed treatments for rare disease.
Applied Clinical Trials
CDER's Rare Disease Cures Accelerator initiative looks to foster a coordinated research approach and methods that can expedite development of drugs to treat some of the 7,000 rare diseases.

Applied Clinical Trials
Concerns for patient safety are raised with the recent surge in new drugs receiving fast-track FDA approval. Jill Wechsler reports.

Applied Clinical Trials
FDA draft guidance for modernizing the approach to clinical trial design for oncology drugs and biologics look to make clinical trials more efficient while maintaining patient safety.
Applied Clinical Trials
A new CDER “knowledge management” approach will see companies submit applications that can be transmitted to experts from multiple disciplines able to assess applications for new drugs and biologics in a timely and efficient manner.

Applied Clinical Trials
Supporting real change in clinical trials is more than just lip service-it’s putting the information out there transparently for all stakeholders to measure and make decisions.

Applied Clinical Trials
Jill Wechsler details new FDA policies to streamline drug development, including the design of “seamless” trials.
Applied Clinical Trials
Rho offers their expertise on why it is important to have a pre-IND meeting to ensure a successful IND.
Advertisement
Applied Clinical Trials
With the EU’s new General Data Protection Regulation coming into force in May this year, the impetus for life sciences firms to cement their data management strategies has increased.
Applied Clinical Trials
An overview of all the drug approvals that were secured in 2017.
Applied Clinical Trials
Under pressure to meet tight deadlines for reviewing and approving a growing volume of applications for new drugs, generics, and medical products, FDA is rejecting submissions that are incomplete or unsatisfactory right from the start.
Applied Clinical Trials
The FDA published a final rule on FDA’s standards for accepting data from clinical investigations for medical devices on February 21.

Applied Clinical Trials
Jill Wechsler details the two chief reasons why clinical trial quality and efficiency has improved in recent years.
Applied Clinical Trials
The FDA's new draft IVD Guidelines will need to be factored into how pharma clinical trial sponsors use IVDs in a clinical trial, study design, timeline for protocol development, IND submission, and study initiation.

Applied Clinical Trials
Now the challenge to FDA and to sponsors is to maintain the high level of support for research, discovery, and regulatory flexibility underpinning these gains, writes Jill Wechsler.

Applied Clinical Trials
In this interview, Austin Speier, VP of Emerging Technologies at Precision for Medicine, will elaborate on these draft guidance documents.
Applied Clinical Trials
The agency is moving to smooth the pathways for orphan drugs, genetic therapies, and other scientific advances to yield more transformative medicines.
Applied Clinical Trials
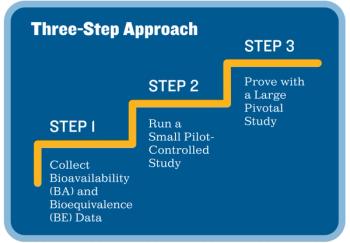
Sponsors can streamline the design and execution of their clinical trials by following this three-step approach.
Applied Clinical Trials
Jill Wechsler talks about biosimilars and the requirement of clinical trials in her recent blog.

Applied Clinical Trials
This article will dive into the details of FDA’s movement in mHealth, analyze FDA’s approach, and assess how this movement impacts the use of mHealth in clinical trial settings.
Applied Clinical Trials
New legislation aims to expand regulatory acceptance of patient data from healthcare systems and observational studies.
Applied Clinical Trials
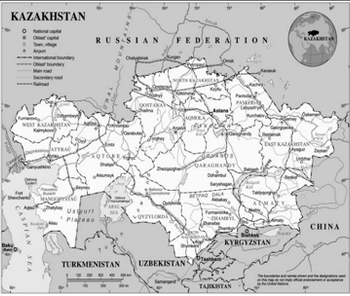
This brief overview of Kazakhstan presented by Vlad Bogin will help you get acquainted with this emerging clinical trial location.
Applied Clinical Trials
There is a critical need to rethink standards of evidence and of the reliability of information used to make regulatory decisions. According to the FDA, this involves placing greater reliance on data from sources outside traditional clinical studies and because of these new tools for collecting the data, the FDA needs to adapt as well.
Advertisement
Advertisement
Trending on Applied Clinical Trials Online
1
ACT Brief: Execution Failures and Upstream Origins, European Data Governance Reshaping CROs, and Phase-Dependent Trial Competition
2
Real-World Evidence in Transition: CRO Adaptation to the European Health Data Space Framework
3
ACT Brief: Sponsor Oversight Capability Gaps, Community-Embedded Trial Partnerships, and Trialblazer Timeline Realism
4
The AI Churn Trap in Life Sciences and How to Break It
5